Polishing the Final Assembly#
After generating the updated consensus from Verkko using revised paths and performing necessary post-processing steps—such as trimming, sorting, and renaming—the assembly is ready for polishing.
Polishing addresses small sequence errors in the assembly. Several established tools exist for this purpose, such as Racon[1] and DeepPolisher[2]. In this tutorial, we focus on a manual curation approach based on the T2T-Ref GitHub repository[3] and the Primate T2T paper[4]. This method uses DeepVariant[5] for variant detection, followed by stringent filtering to retain only true variants. The resulting high-confidence variant set is then applied to generate an error-corrected assembly. The diagram below illustrates all the files that need to be prepared, along with the options recommended in the Primate T2T paper[4].

The polishing workflow consists of the following steps:
Generate Haplotype Specific References
Align ONT, HiFi, and Illumina reads
Create a hybrid alignment (HiFi and Illumina combined)
Run DeepVariant in different modes depending on the reference type
Build HiFi-illumina hybrid meryl Db and get the haploid coverage
Filter variants precisely to retain only true variants
Build a new consensus using the true variant set
(Optional but Recommended) Perform quality checks on the new assembly
Generate Haplotype Specific References#
For diploid genomes, it is necessary to generate haplotype-specific references—for example, hap1.fasta (maternal) and hap2.fasta (paternal). Each haplotype-specific reference should include all autosomes for that haplotype, both sex chromosomes (chrX and chrY), the mitochondrial chromosome (chrM), and any additional accessory chromosomes, if present.
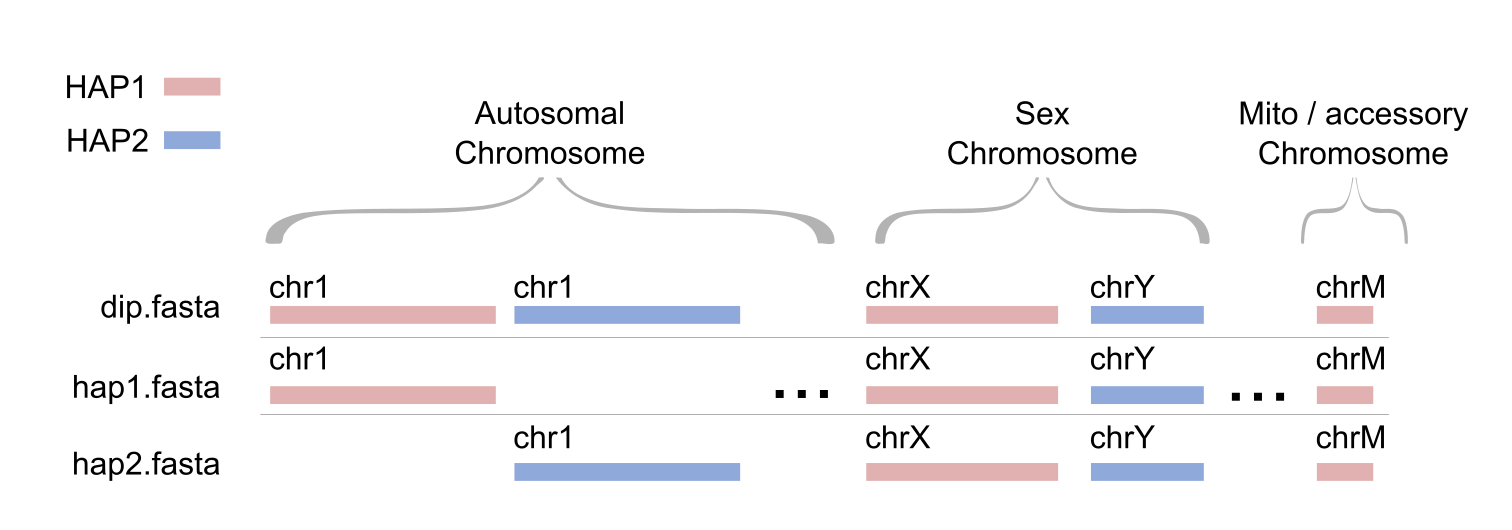
Expected outputs for giraffe genome:
hap1.fasta: autosomes from hap1 + chrX + chrY + chrM
hap2.fasta: autosomes from hap2 + chrX + chrY + chrM
dip.fasta: all chromosomes from hap1 and hap2 + chrX + chrY + chrM
Example extraction using samtools:
# For hap1
samtools faidx dip.fasta chr1_mat chr2_mat .... chr14_mat chrX_mat chrY_pat chrM_mat > hap1.fasta &&
samtools faidx hap1.fasta
# For hap2
samtools faidx dip.fasta chr1_pat chr2_pat .... chr14_pat chrX_mat chrY_pat chrM_mat > hap2.fasta &&
samtools faidx hap2.fasta
Align ONT, HiFi, and Illumina reads#
The ONT reads are aligned to the diploid reference, while the HiFi and Illumina short reads are aligned to both the diploid reference and each haplotype-specific reference. Only primary alignments are retained after filtering. For Illumina read alignments, duplicate reads are removed during the deduplication step.
For ONT and HiFi read alignments to the diploid, hap1, and hap2 genomes, we use winnowmap.
# Indexing and extract kmers before aligning the reads
# by reference
ref={hap1/hap2/dip}
meryl count k=15 ${ref}.fasta output ${ref}.merylDB # add "compress" for homopolymer compression
meryl print greater-than distinct=0.9998 ${ref}.merylDB > ${ref}.repetitive_k15.txt
# Aligning
# by platform and reference
platform={map-ont/map-pb}
reads=/where/to/ONT.1.fastq.gz
array=1 # If you have more than one FASTQ file from the same platform, you will generate multiple BAM files with different name.
winnowmap --MD -W ${ref}.repetitive_k15.txt -ax ${platform} -I12g -y -t$cpus ${ref}.fasta $reads > ${platform}.${ref}.${array}.sam
samtools sort -@$cpus -m2G -O bam -o ${platform}.${ref}.${array}.sort.sam ${platform}.${ref}.${array}.sam
# Merge BAM files before proceeding to the next step.
samtools merge -O bam -@48 ${platform}.${ref}.bam ${platform}.${ref}*sort.bam
samtools index ${platform}.${ref}.bam
# Filter out secondary alignments
samtools view -F0x104 -@12 -hb ${platform}.${ref}.bam > ${platform}.${ref}.pri.bam
samtools index ${platform}.${ref}.pri.bam
For Illumina read alignments to the diploid, hap1, and hap2 genomes, we use bwa.
# Indexing before aligning the reads
ref={hap1/hap2/dip}
bwa index ${ref}.fasta
r1=/where/to/illumina_1.fastq.gz
r2=/where/to/illumina_2.fastq.gz
bwa mem -t $cpu ${ref}.fasta $r1 $r2 > illu.${ref}.sam
samtools fixmate -m -@$cpu illu.${ref}.sam illu.${ref}.fix.bam && rm illu.${ref}.sam
samtools sort -@$cpu -O bam -o illu.${ref}.bam illu.${ref}.fix.bam && rm illu.${ref}.fix.bam
samtools index -@$cpu illu.${ref}.fix.bam
samtools markdup -r -@$cpu illu.${ref}.bam illu.${ref}.dedup.bam && rm illu.${ref}.bam
samtools index -@$cpu illu.${ref}.dedup.bam
# Filter out secondary alignments
samtools view -@$cpu -F0x100 -hb --write-index -o illu.${ref}.dedup.pri.bam illu.${ref}.dedup.bam &&
Outputs are :
ONT/HiFi/Illumina aligned on the dip.fasta ->
ONT.dip.pri.bam/HiFi.dip.pri.bam/illu.dip.pri.bamHiFi/Illumina aligned on the hap1.fasta ->
HiFi.hap1.pri.bam/illu.hap1.pri.bamHiFi/Illumina aligned on the hap2.fasta ->
HiFi.hap2.pri.bam/illu.hap2.pri.bam
Create a hybrid alignment (HiFi and Illumina combined)#
To run DeepVariant in HYBRID_PACBIO_ILLUMINA mode[6] in the next step, the HiFi and Illumina short-read alignments must be merged into a single BAM file for each reference (diploid, hap1, and hap2).
# for dip
samtools merge hybrid.dip.pri.bam HiFi.dip.pri.bam illu.dip.dedup.pri.bam &&
samtools index hybrid.dip.pri.bam
# for hap1
samtools merge hybrid.hap1.pri.bam HiFi.hap1.pri.bam illu.hap1.dedup.pri.bam &&
samtools index hybrid.hap1.pri.bam
# for hap2
samtools merge hybrid.hap2.pri.bam HiFi.hap2.pri.bam illu.hap2.dedup.pri.bam &&
samtools index hybrid.hap2.pri.bam
Outputs are
hybrid.{dip/hap1/hap2}.pri.bamand their indexs (.bai)
Run DeepVariant in different modes depending on the reference type#
Note that when using BAM files aligned to the diploid reference (dip.fasta), a mapping quality (MAPQ) threshold of 0 was applied. For BAM files aligned to haplotype-specific references (hap1.fasta or hap2.fasta), a MAPQ threshold of -1 was used. Additionally, ensure that the DeepVariant mode corresponds to the read type: use ONT mode for ONT reads and HYBRID_PACBIO_ILLUMINA mode for combined HiFi and Illumina reads. We employ a script from the T2T-polishing GitHub repository[3] (download link). This script requires five inputs, in order: the reference genome, the BAM file, the DeepVariant mode, the sample name, and the mapping quality threshold.
For ONT aligned on dip.fasta
_submit_deepvariant_with_minqual.sh dip.fasta ONT.dip.pri.bam ONT_R104 giraffe 0
For hybrid aligned on dip.fasta
_submit_deepvariant_with_minqual.sh dip.fasta hybrid.dip.pri.bam HYBRID_PACBIO_ILLUMINA giraffe 0
For hybrid aligned on hap1/2.fasta
# for hap1
_submit_deepvariant_with_minqual.sh hap1.fasta hybrid.hapa.pri.bam HYBRID_PACBIO_ILLUMINA giraffe -1
# for hap2
_submit_deepvariant_with_minqual.sh hap2.fasta hybrid.hap2.pri.bam HYBRID_PACBIO_ILLUMINA giraffe -1
Outputs are
ONT.dip.vcfhybrid.dip.vcfhybrid.hap1.vcfhybrid.hap2.vcf
Build HiFi-illumina hybrid meryl Db and get the haploid coverage#
The hybrid k-mer databases from HiFi and Illumina reads, along with the estimated coverage for each haplotype, are required for the next step. We use meryl with a k-mer size of 31 to generate k-mer databases separately from HiFi and Illumina reads using meryl[7]. Unique k-mers present exclusively in one database are then filtered, and the two databases are merged to create the hybrid k-mer database.
# Generating meryl DB
platform={hifi/Illumina}
MERQURY=/where/to/merqury/installed
faDir=/where/to/fastqs
realpath $faDir > ${platform}.fofn
$MERQURY/_submit_build.sh 31 ${platform}.fofn ${platform}_meryl
# Filtering unique kmers
meryl greater-than 1 illumina_meryl.k31.meryl output illumina_meryl.k31.gt1.meryl
meryl greater-than 1 hifi_meryl.k31.meryl output hifi_meryl.k31.gt1.meryl
# Merging HiFi and illumina meryl DB
meryl union-sum illumina_meryl.k31.gt1.meryl hifi_meryl.k31.gt1.meryl output illu.hifi.hybrid.k31.meryl
The haploid coverage can be estimated using the hybrid k-mer database generated above.
meryl histogram illu.hifi.hybrid.k31.meryl > hybrid.k31.kmer_freq.hist
By plotting a histogram of k-mer multiplicity versus k-mer counts, the coverage distribution becomes apparent. In diploid genomes, this histogram typically displays two prominent peaks: the first peak represents haploid coverage, while the second corresponds to diploid coverage. For example, in the giraffe genome, the haploid coverage peak occurs around 68, which will be used in subsequent variant filtering steps.
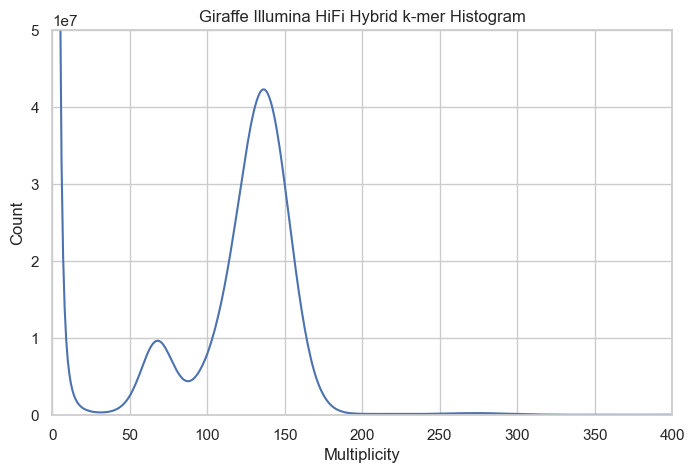
Outputs are
illu.hifi.hybrid.k31.merylpeak : estimated coverage for one haplotype
Filter variants precisely to retain only true variants#
We obtained four VCF files as described above, which we then merged. To filter out less confident variant calls, we used a script from the T2T-Ref pipeline. Within this script, variants with a quality score below 00 were filtered out. Finally, Merfin was applied to remove variants overlapping error-prone k-mers.
fa=dip.fasta
ver=1.0 # version you want
ont_to_dip=ONT.dip.pri.vcf
hybrid_to_dip=ONT.dip.pri.vcf
hybrid_to_hap1=ONT.dip.pri.vcf
hybrid_to_hap2=ONT.dip.pri.vcf
hybridkmer=illu.hifi.hybrid.k31.meryl # from step5
peak=68 # from step5
mkdir -p polishing_variants && cd polishing_variants
sh snv_candidates.sh $fa $ver $ont_to_dip $hybrid_to_dip $hybrid_to_hap1 $hybrid_to_hap2 $hybridkmer $peak
Output is
snv_candidates.merfin-loose.vcf.gz
You can filter out specific contigs or regions that you want to exclude. In this case, we excluded the chrM and telomeric regions from each contig.
tel="assembly_dip.telomere.bed" # from getT2T function in the verkko-fillet
bcftools view -T ^${tel} -o snv_candidates.merfin-loose.exc_tel.vcf -O v snv_candidates.merfin-loose.vcf.gz
bgzip snv_candidates.merfin-loose.exc_tel.vcf
tabix -p vcf snv_candidates.merfin-loose.exc_tel.vcf.gz
grep -v -e "random" -e "rDNA" -e "chrM" ${ori_fa}.fai | awk '{print $1"\t"0"\t"$2}' > main_chrom.bed
bcftools view -R main_chrom.bed $finalVCF -Oz -o snv_candidates.merfin-loose.mainChr.vcf.gz
tabix -p vcf snv_candidates.merfin-loose.mainChr.vcf.gz
Output is
snv_candidates.merfin-loose.exc_tel.vcf.gz
Build a new consensus using the true variant set#
Once the confident variant call set is ready. The new assembly could be generated using bcftools consensus.
bcftools consensus -H1 --chain dip_to_dipPolished.chain -f $ori_fa snv_candidates.merfin-loose.exc_tel.vcf.gz > dip.polished.fasta
(Optional but Recommended) Perform quality checks on the new assembly#
Once the polishing step is complete, the assembly is ready for downstream analyses or mapping. However, it is important to assess the assembly quality before proceeding. Below are some widely used assembly evaluation tools:
References#
Here are the papers and links referenced in this document.