Recovering Telomere-to-Telomere Contigs#
You need Verkko version > 0.1.12 for this tutorial
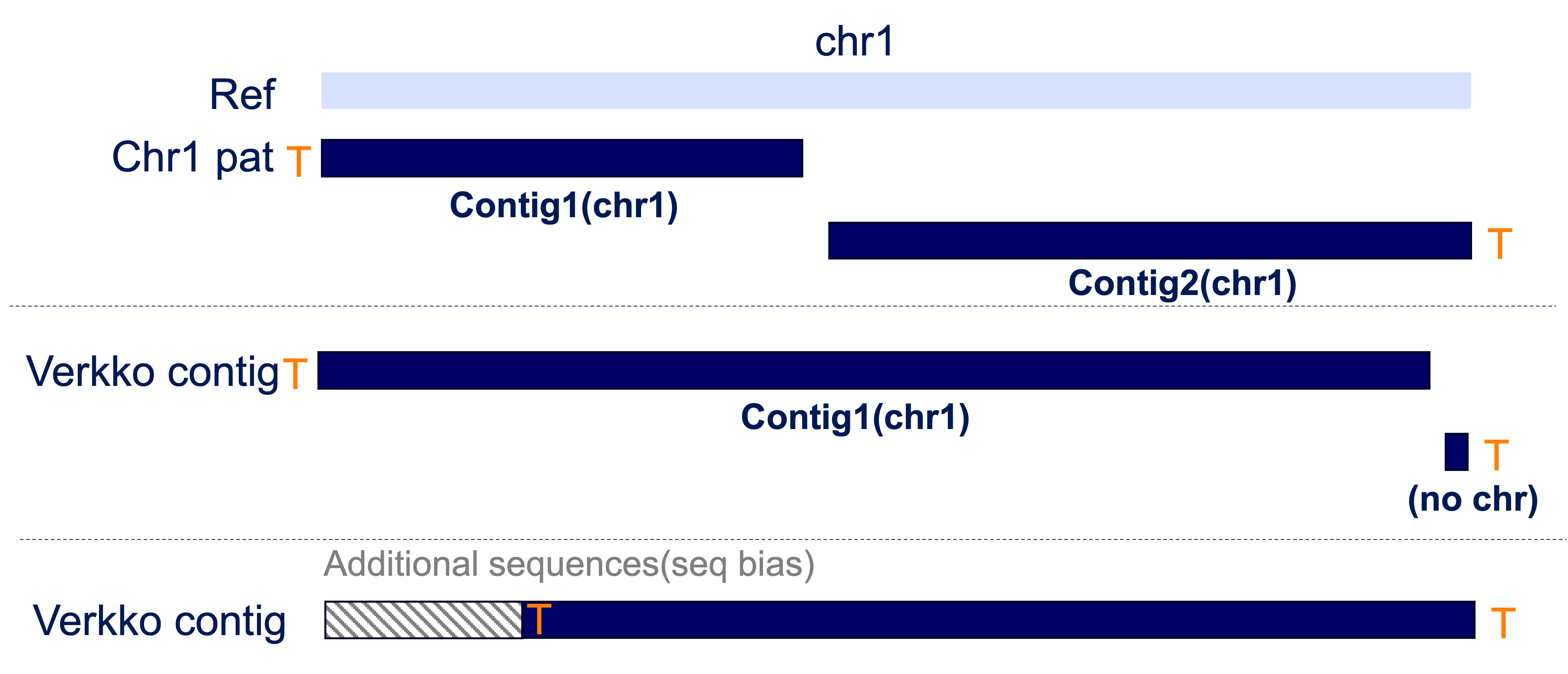
After building the Verkko assembly, we can check how many contigs have telomeres at both ends to determine if the assembly is truly T2T (telomere-to-telomere). Ideally, most contigs should have telomeres, but don’t worry if some do not. In this section, we introduce several cases where you can recover T2T contigs.
We will cover three different scenarios:
Case 1: A single contig was mistakenly split into two, but chromosome assignment information is available.
Case 2: A single contig was mistakenly split into two, but chromosome assignment information is missing for the shorter contig.
Case 3: Artificial sequences were added beyond the real telomere, preventing telomere detection at the ends.
image_path = os.path.dirname(vf.__file__) + "/data/test_giraffe/fig/missingTel.jpg"
display(Image(filename=image_path,width=1000))
%load_ext autoreload
%autoreload 2
import sys
import importlib
import pandas as pd
import time
import os
from IPython.display import Image, display
pd.set_option('mode.chained_assignment', None)
import matplotlib.pyplot as plt
from itables import init_notebook_mode
init_notebook_mode(all_interactive=True)
import warnings
import session_info
# Suppress FutureWarnings
warnings.simplefilter(action='ignore', category=FutureWarning)
import sys
import tqdm
from importlib import reload
print(os.getcwd())
sys.path.append('/data/Phillippy/projects/giraffeT2T/assembly/script/verkko-fillet/src')
import verkkofillet as vf
vf = reload(vf)
/path/to/your/folder/script/test_notebooks/basics
verkko-fillet version:0.1.19
verkko-fillet version:0.1.19
verkkoDir = '/path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic'
obj = vf.pp.read_Verkko(verkkoDir)
# print(obj.verkko_fillet_dir)
os.chdir(obj.verkko_fillet_dir)
The Verkko fillet target directory already exists: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet
If you didn't mean this, please set another directory or for overwirting, please use force= True
Lock the original Verkko folder to prevent it from being modified.
[lock_original_folder] Command executed successfully!
change working directory: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet
Path file loaded successfully.
scfmap file loaded successfully.
Reading assembly.homopolymer-compressed.noseq.gfa
Reading assembly.colors.csv
Node information is stored in obj.node
fileName = "/path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet/verkko-fillet_gapfilled.230519.pkl"
obj = vf.pp.load_Verkko(fileName)
load verkko fllet obj from <- /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet/verkko-fillet_gapfilled.230519.pkl
obj = vf.pp.findGaps(obj)
45 gaps were found -> obj.gaps
map_file= "/path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_legacy/chromosome.map"
obj = vf.pp.readChr(obj, map_file, sire = "pat", dam = "mat")
The chromosome infomation was stored in obj.stats
vf.pl.contigPlot(obj,plot_height = 4 , plot_width = 2)
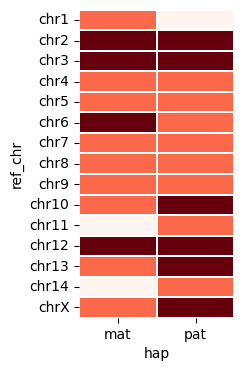
Here, we have three contigs missing one of telomeres: chr11_mat, chr14_mat, and chr1_pat.
Finding broken contigs#
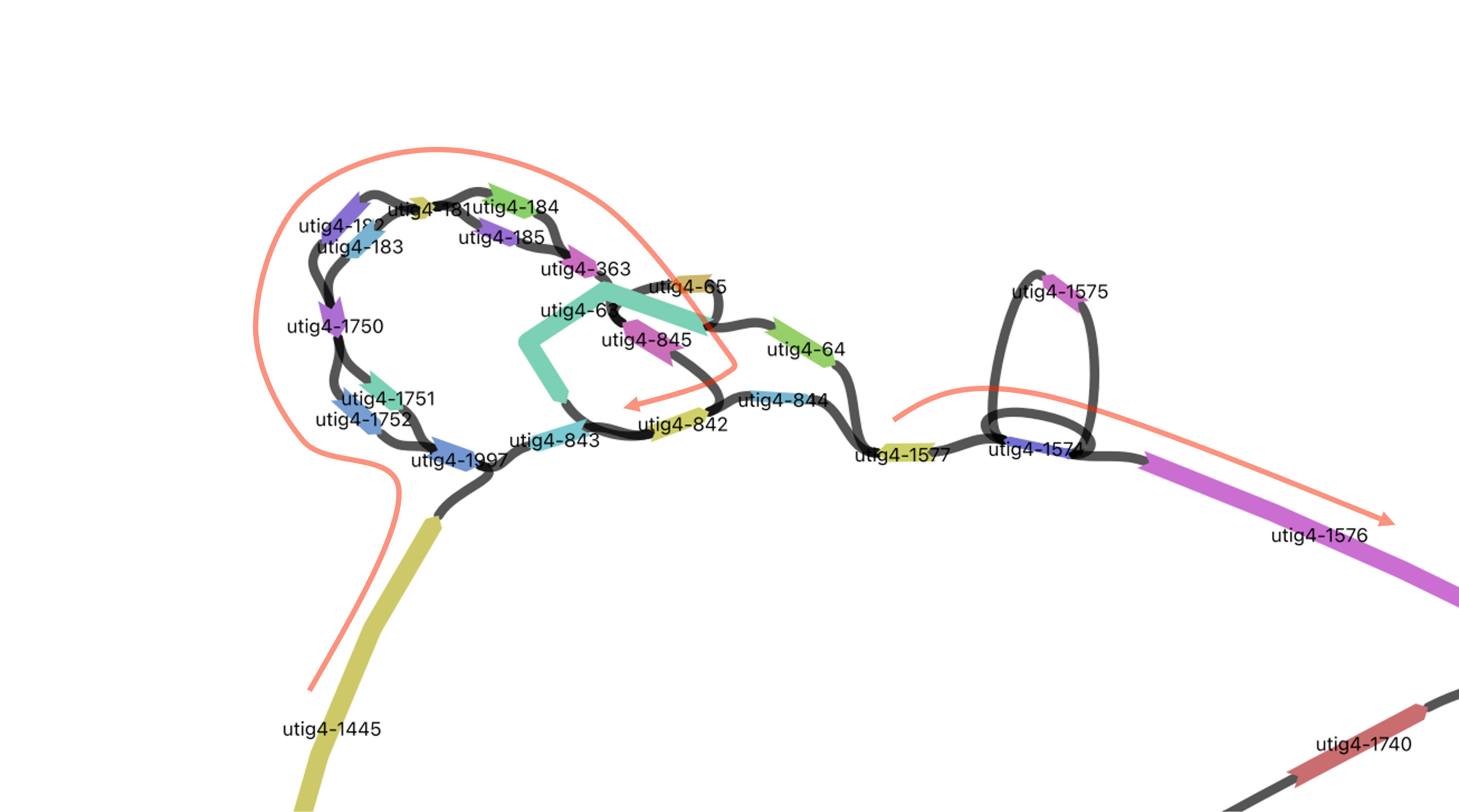
Using the vf.pp.detectBrokenContigs() function, you can identify chromosomes that have more than two contigs from the same haplotype. This indicates that Verkko generated two distinct contigs, each long enough to be assigned to a chromosome, even though they originally belonged to a single contig.
In the giraffe genome, this issue occurs in the complex repeat and loop region in the middle of chr1 for the paternal haplotype.
vf.pp.detectBrokenContigs(obj)
Warning: the following chromosomes have more than one contig:
ref_chr hap_verkko contig
0 chr1 sire 2
obj.stats.loc[obj.stats['ref_chr'] == 'chr1']
| contig | ref_chr | contig_len | ref_chr_len | hap | old_chr | completeness | hap_verkko | t2tStat | |
|---|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
brokenContigs_chr1_pat_contigName = ['sire_compressed.k31.hapmer-0000240','sire_compressed.k31.hapmer-0000243']
obj.scfmap[obj.scfmap['contig'].isin(brokenContigs_chr1_pat_contigName)]
| contig | pathName | |
|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
If we examine the paths of each contig, we can see that the breakage occurs between utig-842 and utig-1577.
brokenContigs_chr1_pat_pathName=['sire_compressed.k31.hapmer_from_utig4-1445','sire_compressed.k31.hapmer_from_utig4-1859']
obj.paths.loc[obj.paths['name'].isin(brokenContigs_chr1_pat_pathName)]
| name | path | assignment | |
|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
image_path = os.path.dirname(vf.__file__) + "/data/test_giraffe/fig/pat_chr1_brokenContig.png"
display(Image(filename=image_path,width=1000))
We are going to connect these two paths by inserting a new gap of 5 kb. Before doing so, here is the current state of obj.gaps:
obj.gaps.tail(2)
| gapId | name | gaps | notes | fixedPath | startMatch | endMatch | finalGaf | done | |
|---|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
The vf.pp.connectContigs() function is used to merge contigs that are separated by gaps. This function takes the following arguments:
contig: The contig to be connected.contig_to: The target contig to which it will be connected.at: The position where the contig should be connected to contig_to.flip: Set to True if you want to flip the contig.fix_path: Set to True if you want to updateobj.path. We don’t recommend update this.
In this example, we will connect sire_compressed.k31.hapmer_from_utig4-1445 to the left side (beginning) of sire_compressed.k31.hapmer_from_utig4-1859 using a new gap named [N5000N:insertGap], without flipping the contig.
You can directly check how the new gap has been added to the obj.gaps database.
If you accidentally add incorrect gap information, you can remove it using the vf.pp.deleteGap function, as shown below. This example demonstrates how to create a new gap, remove an incorrect attempt, and then correct it.
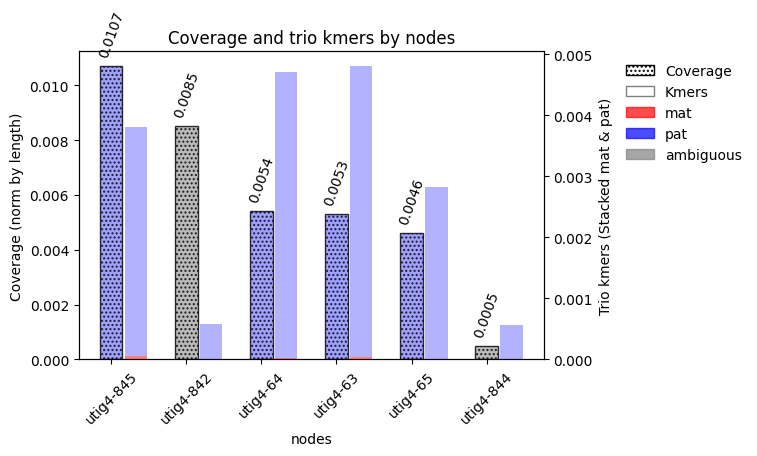
nodlist = 'utig4-63, utig4-64, utig4-65, utig4-842, utig4-844, utig4-845'
node_list = nodlist.replace(" ", "").split(",")
node_list
['utig4-63', 'utig4-64', 'utig4-65', 'utig4-842', 'utig4-844', 'utig4-845']
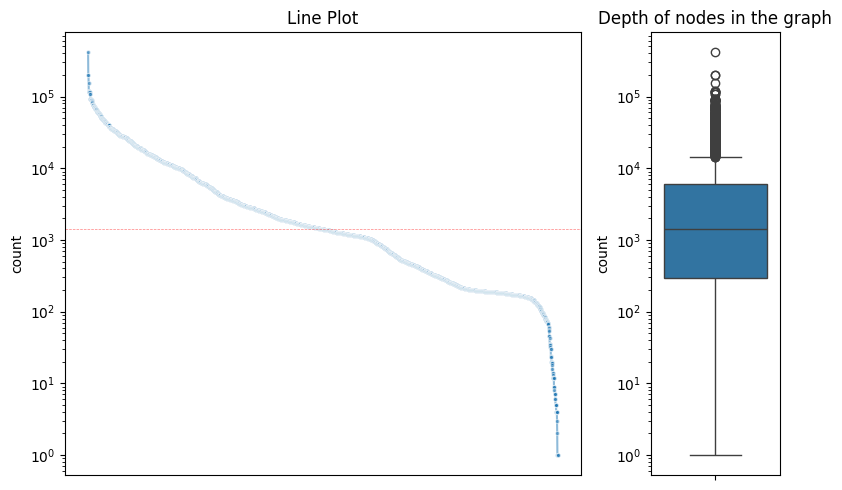
obj = vf.pp.calNodeDepth(obj)
Counting the frequency of each node in the paths...
Median normalized by length: 0.007214537059447785
Median of haplotype specific nodes normalized by length : 0.005721821461611718
Plotting the depth of nodes in the graph...
File figs/nodeDepth.png already exists
Please remove the file or change the name
obj.node.loc[obj.node['node'].isin(node_list),:]
| node | len | mat | pat | mat:pat | color | ont_cov | hifi_cov | hap | count | norm_len | cov | cov_hap | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
vf.pl.barCovKmer(obj, node_list, save=True,figName = "/data/Phillippy/projects/giraffeT2T/assembly/script/giraffe_codes/fig/chr1_pat_brocken.pdf")
INFO:fontTools.subset:maxp pruned
INFO:fontTools.subset:cmap pruned
INFO:fontTools.subset:kern dropped
INFO:fontTools.subset:post pruned
INFO:fontTools.subset:FFTM dropped
INFO:fontTools.subset:GPOS pruned
INFO:fontTools.subset:GSUB pruned
INFO:fontTools.subset:glyf pruned
INFO:fontTools.subset:Added gid0 to subset
INFO:fontTools.subset:Added first four glyphs to subset
INFO:fontTools.subset:Closing glyph list over 'MATH': 42 glyphs before
INFO:fontTools.subset:Glyph names: ['.notdef', '.null', 'C', 'K', 'S', 'T', 'a', 'ampersand', 'b', 'c', 'd', 'e', 'eight', 'five', 'four', 'g', 'h', 'hyphen', 'i', 'k', 'l', 'm', 'n', 'nonmarkingreturn', 'o', 'one', 'p', 'parenleft', 'parenright', 'period', 'r', 's', 'seven', 'six', 'space', 't', 'three', 'two', 'u', 'v', 'y', 'zero']
INFO:fontTools.subset:Glyph IDs: [0, 1, 2, 3, 9, 11, 12, 16, 17, 19, 20, 21, 22, 23, 24, 25, 26, 27, 38, 46, 54, 55, 68, 69, 70, 71, 72, 74, 75, 76, 78, 79, 80, 81, 82, 83, 85, 86, 87, 88, 89, 92]
INFO:fontTools.subset:Closed glyph list over 'MATH': 48 glyphs after
INFO:fontTools.subset:Glyph names: ['.notdef', '.null', 'C', 'K', 'S', 'T', 'a', 'ampersand', 'b', 'c', 'd', 'e', 'eight', 'five', 'four', 'g', 'h', 'hyphen', 'i', 'k', 'l', 'm', 'n', 'nonmarkingreturn', 'o', 'one', 'p', 'parenleft', 'parenright', 'period', 'r', 's', 'seven', 'six', 'space', 't', 'three', 'two', 'u', 'uni239B', 'uni239C', 'uni239D', 'uni239E', 'uni239F', 'uni23A0', 'v', 'y', 'zero']
INFO:fontTools.subset:Glyph IDs: [0, 1, 2, 3, 9, 11, 12, 16, 17, 19, 20, 21, 22, 23, 24, 25, 26, 27, 38, 46, 54, 55, 68, 69, 70, 71, 72, 74, 75, 76, 78, 79, 80, 81, 82, 83, 85, 86, 87, 88, 89, 92, 3506, 3507, 3508, 3509, 3510, 3511]
INFO:fontTools.subset:Closing glyph list over 'GSUB': 48 glyphs before
INFO:fontTools.subset:Glyph names: ['.notdef', '.null', 'C', 'K', 'S', 'T', 'a', 'ampersand', 'b', 'c', 'd', 'e', 'eight', 'five', 'four', 'g', 'h', 'hyphen', 'i', 'k', 'l', 'm', 'n', 'nonmarkingreturn', 'o', 'one', 'p', 'parenleft', 'parenright', 'period', 'r', 's', 'seven', 'six', 'space', 't', 'three', 'two', 'u', 'uni239B', 'uni239C', 'uni239D', 'uni239E', 'uni239F', 'uni23A0', 'v', 'y', 'zero']
INFO:fontTools.subset:Glyph IDs: [0, 1, 2, 3, 9, 11, 12, 16, 17, 19, 20, 21, 22, 23, 24, 25, 26, 27, 38, 46, 54, 55, 68, 69, 70, 71, 72, 74, 75, 76, 78, 79, 80, 81, 82, 83, 85, 86, 87, 88, 89, 92, 3506, 3507, 3508, 3509, 3510, 3511]
INFO:fontTools.subset:Closed glyph list over 'GSUB': 48 glyphs after
INFO:fontTools.subset:Glyph names: ['.notdef', '.null', 'C', 'K', 'S', 'T', 'a', 'ampersand', 'b', 'c', 'd', 'e', 'eight', 'five', 'four', 'g', 'h', 'hyphen', 'i', 'k', 'l', 'm', 'n', 'nonmarkingreturn', 'o', 'one', 'p', 'parenleft', 'parenright', 'period', 'r', 's', 'seven', 'six', 'space', 't', 'three', 'two', 'u', 'uni239B', 'uni239C', 'uni239D', 'uni239E', 'uni239F', 'uni23A0', 'v', 'y', 'zero']
INFO:fontTools.subset:Glyph IDs: [0, 1, 2, 3, 9, 11, 12, 16, 17, 19, 20, 21, 22, 23, 24, 25, 26, 27, 38, 46, 54, 55, 68, 69, 70, 71, 72, 74, 75, 76, 78, 79, 80, 81, 82, 83, 85, 86, 87, 88, 89, 92, 3506, 3507, 3508, 3509, 3510, 3511]
INFO:fontTools.subset:Closing glyph list over 'glyf': 48 glyphs before
INFO:fontTools.subset:Glyph names: ['.notdef', '.null', 'C', 'K', 'S', 'T', 'a', 'ampersand', 'b', 'c', 'd', 'e', 'eight', 'five', 'four', 'g', 'h', 'hyphen', 'i', 'k', 'l', 'm', 'n', 'nonmarkingreturn', 'o', 'one', 'p', 'parenleft', 'parenright', 'period', 'r', 's', 'seven', 'six', 'space', 't', 'three', 'two', 'u', 'uni239B', 'uni239C', 'uni239D', 'uni239E', 'uni239F', 'uni23A0', 'v', 'y', 'zero']
INFO:fontTools.subset:Glyph IDs: [0, 1, 2, 3, 9, 11, 12, 16, 17, 19, 20, 21, 22, 23, 24, 25, 26, 27, 38, 46, 54, 55, 68, 69, 70, 71, 72, 74, 75, 76, 78, 79, 80, 81, 82, 83, 85, 86, 87, 88, 89, 92, 3506, 3507, 3508, 3509, 3510, 3511]
INFO:fontTools.subset:Closed glyph list over 'glyf': 48 glyphs after
INFO:fontTools.subset:Glyph names: ['.notdef', '.null', 'C', 'K', 'S', 'T', 'a', 'ampersand', 'b', 'c', 'd', 'e', 'eight', 'five', 'four', 'g', 'h', 'hyphen', 'i', 'k', 'l', 'm', 'n', 'nonmarkingreturn', 'o', 'one', 'p', 'parenleft', 'parenright', 'period', 'r', 's', 'seven', 'six', 'space', 't', 'three', 'two', 'u', 'uni239B', 'uni239C', 'uni239D', 'uni239E', 'uni239F', 'uni23A0', 'v', 'y', 'zero']
INFO:fontTools.subset:Glyph IDs: [0, 1, 2, 3, 9, 11, 12, 16, 17, 19, 20, 21, 22, 23, 24, 25, 26, 27, 38, 46, 54, 55, 68, 69, 70, 71, 72, 74, 75, 76, 78, 79, 80, 81, 82, 83, 85, 86, 87, 88, 89, 92, 3506, 3507, 3508, 3509, 3510, 3511]
INFO:fontTools.subset:Retaining 48 glyphs
INFO:fontTools.subset:head subsetting not needed
INFO:fontTools.subset:hhea subsetting not needed
INFO:fontTools.subset:maxp subsetting not needed
INFO:fontTools.subset:OS/2 subsetting not needed
INFO:fontTools.subset:hmtx subsetted
INFO:fontTools.subset:cmap subsetted
INFO:fontTools.subset:fpgm subsetting not needed
INFO:fontTools.subset:prep subsetting not needed
INFO:fontTools.subset:cvt subsetting not needed
INFO:fontTools.subset:loca subsetting not needed
INFO:fontTools.subset:post subsetted
INFO:fontTools.subset:gasp subsetting not needed
INFO:fontTools.subset:MATH subsetted
INFO:fontTools.subset:GDEF subsetted
INFO:fontTools.subset:GPOS subsetted
INFO:fontTools.subset:GSUB subsetted
INFO:fontTools.subset:name subsetting not needed
INFO:fontTools.subset:glyf subsetted
INFO:fontTools.subset:head pruned
INFO:fontTools.subset:OS/2 Unicode ranges pruned: [0]
INFO:fontTools.subset:OS/2 CodePage ranges pruned: [0]
INFO:fontTools.subset:glyf pruned
INFO:fontTools.subset:GDEF pruned
INFO:fontTools.subset:GPOS pruned
INFO:fontTools.subset:GSUB pruned
INFO:fontTools.subset:name pruned
File /data/Phillippy/projects/giraffeT2T/assembly/script/giraffe_codes/fig/chr1_pat_brocken.pdf saved
path1 = "sire_compressed.k31.hapmer_from_utig4-1445" # from
path2 = "sire_compressed.k31.hapmer_from_utig4-1859" # to
gap = "[N5000N:insertGap]"
at = "right" # Wrong orientation for testing
flip = False
fix_path = False
obj = vf.pp.connectContigs(obj, contig= path1, contig_to= path2, at = at, gap = gap, flip = flip, fix_path = fix_path)
obj.gaps.tail(2)
Connected sire_compressed.k31.hapmer_from_utig4-1445 to sire_compressed.k31.hapmer_from_utig4-1859 at right with flip False
sire_compressed.k31.hapmer_from_utig4-1445 was merged to sire_compressed.k31.hapmer_from_utig4-1859 in obj.paths
sire_compressed.k31.hapmer_from_utig4-1445 was replaced with sire_compressed.k31.hapmer_from_utig4-1859 in obj.gaps
New gap was added to obj.gaps with gapId gapid_45
| gapId | name | gaps | notes | fixedPath | startMatch | endMatch | finalGaf | done |
|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
gapId = 'gapid_45'
obj = vf.pp.deleteGap(obj, gapId)
path1 = "sire_compressed.k31.hapmer_from_utig4-1445" # from
path2 = "sire_compressed.k31.hapmer_from_utig4-1859" # to
gap = "[N5000N:insertGap]"
at = "left"
flip = False
fix_path = False
obj = vf.pp.connectContigs(obj, contig= path1, contig_to= path2, at = at, gap = gap, flip = flip, fix_path = fix_path)
obj.gaps.tail(2)
Connected sire_compressed.k31.hapmer_from_utig4-1445 to sire_compressed.k31.hapmer_from_utig4-1859 at left with flip False
sire_compressed.k31.hapmer_from_utig4-1445 was merged to sire_compressed.k31.hapmer_from_utig4-1859 in obj.paths
sire_compressed.k31.hapmer_from_utig4-1445 was replaced with sire_compressed.k31.hapmer_from_utig4-1859 in obj.gaps
New gap was added to obj.gaps with gapId gapid_45
| gapId | name | gaps | notes | fixedPath | startMatch | endMatch | finalGaf | done |
|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
Finding counterpart haplotypes’ chromosome for the broken short nodes#
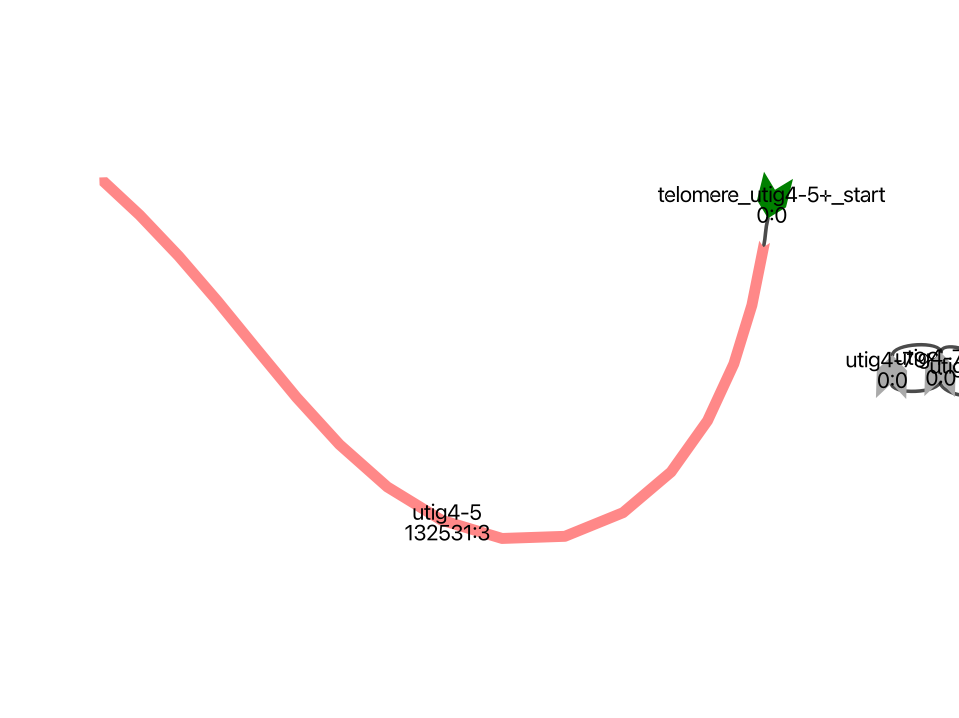
We also have a node that wasn’t assigned to any chromosome due to its short length and lack of connections to other contigs.
In this case, we will align this node to the sequences of other nodes, excluding self-alignment, to identify a corresponding node from the other haplotype. By finding a node from the other haplotype within an intact contig that has a chromosome assignment, we can determine the correct chromosome for this unassigned node and then connect it to the main contig.
image_path = os.path.dirname(vf.__file__) + "/data/test_giraffe/fig/utig4-5_mat.png"
display(Image(filename=image_path,width=500))
Before aligning the node, we need to generate a FASTA file from the graph (.gfa) file. You can use the following command to generate the FASTA file from the graph file:
%%time
vf.tl.gfaToFasta(gfa = "assembly.homopolymer-compressed.gfa", out_fasta=None)
Converting assembly.homopolymer-compressed.gfa to FASTA format
assembly.homopolymer-compressed.fasta already exists
CPU times: user 317 µs, sys: 318 µs, total: 635 µs
Wall time: 1.07 ms
If you have node’s fasta file, you can use it as input fasta file. Otherwise, you can use the output fasta file from the above command to pairwise align the contigs.
%%time
vf.tl.mapBetweenNodes(query = "graphAlignment/utig4-5.fa", threads=50, out=None, showOnly=False,
working_directory="chromosome_assignment")
aligning assembly.homopolymer-compressed.fasta to graphAlignment/utig4-5.fa
Reference: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet/assembly.homopolymer-compressed.fasta
Query: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet/graphAlignment/utig4-5.fa
Output: assembly.homopolymer-compressed_vs_utig4-5.mashmap.out
Indexing reference fasta file
[mapBetNodes] Command executed successfully!
[convertRefName] Command executed successfully!
CPU times: user 7.23 ms, sys: 9.52 ms, total: 16.8 ms
Wall time: 4min 35s
Once the FASTA file is generated, we can determine which node in the graph is the best match for the query sequence.
In this example, the utig4-5 (maternal) node was mapped to the paternal node utig4-1804 and the node in the chr14_pat contig. When examining the chr14, we observe clusters of rDNA nodes at the end of the contig for both haplotypes. However, the paternal contig has an additional node with a telomere node, which indicates the correct chromosome assignment for this sequence.
vf.pl.nodeMashmapBlockSize(mashmap_out = "chromosome_assignment/assembly.homopolymer-compressed_vs_utig4-5.mashmap.out",
node = "utig4-5",
width = 5,
height = 2, figName = "/data/Phillippy/projects/giraffeT2T/assembly/script/giraffe_codes/fig/utig4-5_mappting.pdf")
File /data/Phillippy/projects/giraffeT2T/assembly/script/giraffe_codes/fig/utig4-5_mappting.pdf already exists
Please remove the file or change the name
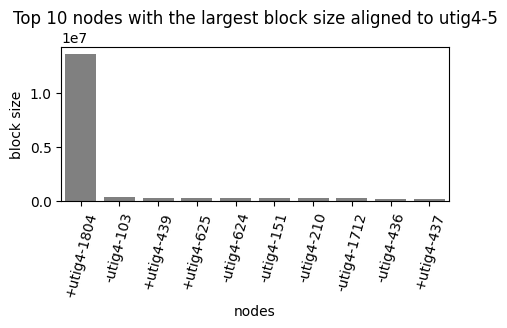
image_path = os.path.dirname(vf.__file__) + "/data/test_giraffe/fig/chr14.jpg"
display(Image(filename=image_path,width=1200))
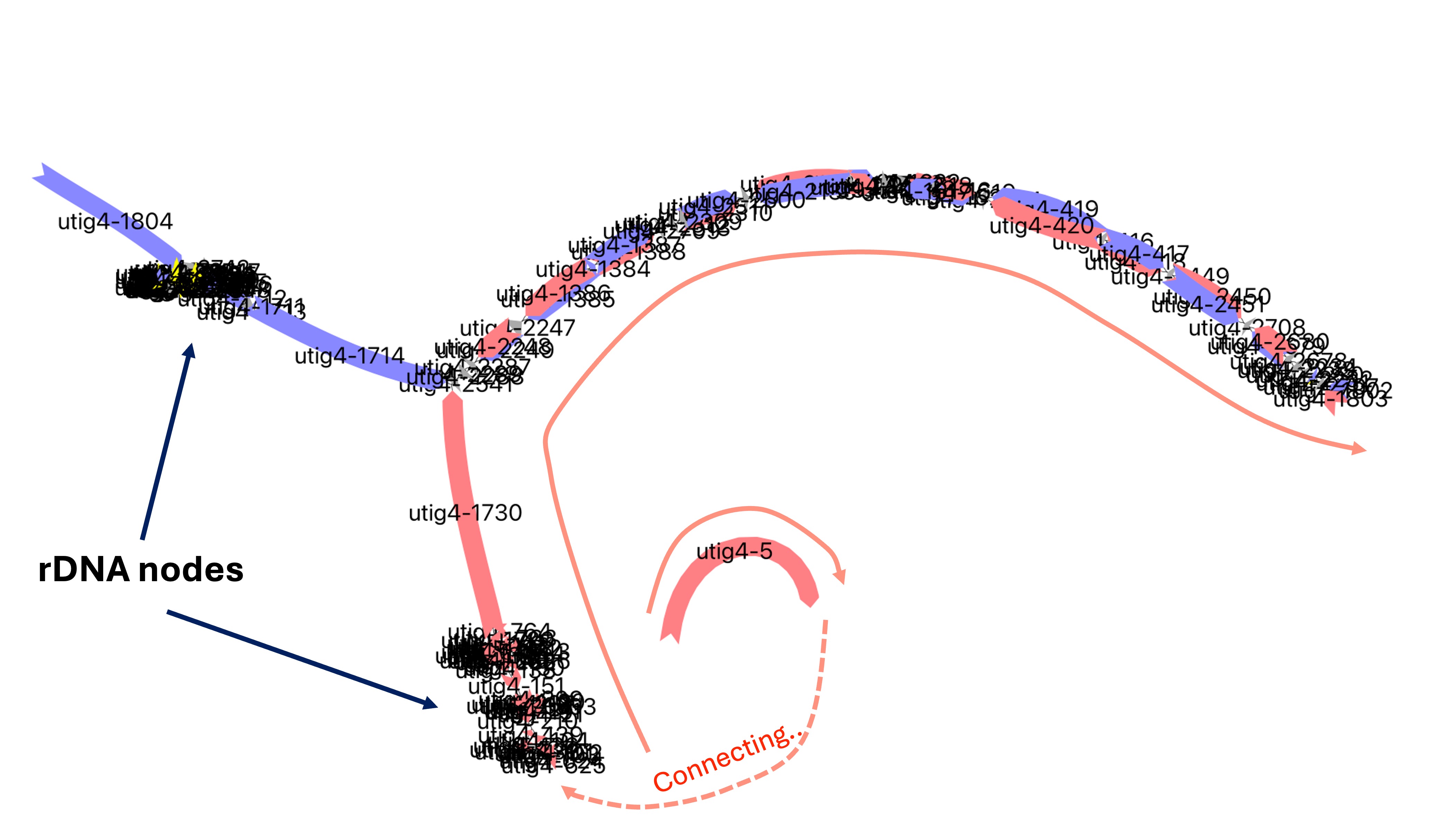
vf.pp.highlight_nodes(obj, node = "utig4-5+")
| name | path |
|---|---|
| haplotype2_from_utig4-5 | utig4-5+ |
vf.pp.highlight_nodes(obj, node = "utig4-1730+")
| name | path |
|---|---|
| dam_compressed.k31.hapmer_from_utig4-1730 | utig4-436-,utig4-439+,utig4-210-,[N572862N:tangle],utig4-151-,[N685960N:tangle],utig4-1730+,utig4-2541+,utig4-2288-,utig4-2287+,utig4-2248-,utig4-2247+,utig4-1386-,utig4-1384+,utig4-1388+,utig4-2709+,utig4-2313-,utig4-2309-,utig4-2310+,utig4-2500+,utig4-2138-,utig4-2137+,utig4-2140+,utig4-2532+,utig4-1621-,utig4-1618-,utig4-1616+,utig4-1619+,utig4-2531-,utig4-420-,utig4-416-,utig4-418+,utig4-2449-,utig4-2450+,utig4-2708-,utig4-2680-,utig4-2678-,utig4-2233-,utig4-2230-,utig4-2231+,utig4-2707+,utig4-1803- |
Since the orientation is the same, we can connect the nodes without flipping.
path1 = "haplotype2_from_utig4-5" # from
path2 = "dam_compressed.k31.hapmer_from_utig4-1730" # to
gap = "[N5000N:insertGap]"
at = "left" # or "right" at path1
flip = False # or True of path2
fix_path = False # if true, the obj.paths will be updated
obj = vf.pp.connectContigs(obj, contig= path1, contig_to= path2, at = at, gap = gap, flip = flip, fix_path = fix_path)
obj.gaps.tail(2)
Connected haplotype2_from_utig4-5 to dam_compressed.k31.hapmer_from_utig4-1730 at left with flip False
haplotype2_from_utig4-5 was merged to dam_compressed.k31.hapmer_from_utig4-1730 in obj.paths
haplotype2_from_utig4-5 was replaced with dam_compressed.k31.hapmer_from_utig4-1730 in obj.gaps
New gap was added to obj.gaps with gapId gapid_45
| gapId | name | gaps | notes | fixedPath | startMatch | endMatch | finalGaf | done |
|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
Finding interal telomere#
If you use HiFi and Dorado reads to build consensus sequences, there is a chance that ONT data extends beyond the true telomere sequences.
In this case, you can use the following functions to identify internal telomere sequences and confirm this by comparing the telomere percentage between internal telomere regions and the telomere sequences at the end of the contig.
Additionally, you can analyze the composition of HiFi and ONT reads at the beginning of the contig—especially if it appears to be an artifact—to check for any sequencing platform bias.
vf.tl.detect_internal_telomere(obj)
intra_telo, tel = vf.pp.find_intra_telo(obj)
intra_telo
Finding the internal telomeres in the assembly...
Reading file: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet/internal_telomere/assembly_1/assembly.windows
Number of internal telomeres: 3
File saved: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet/internal_telomere/assembly_1/assembly.windows.loc_from_end_15000.teloPerct_0.5.telolen_0.stats.tsv
| contig | internal-left | internal-right | non-internal-left | non-internal-right | problem | ref_chr | contig_len | ref_chr_len | hap | old_chr | completeness | hap_verkko | t2tStat |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
vf.pl.percTel(intra_telo, showContig = ['ref_chr','hap'])
File figs/intra_telo.heatmap.png already exists
Please remove the file or change the name
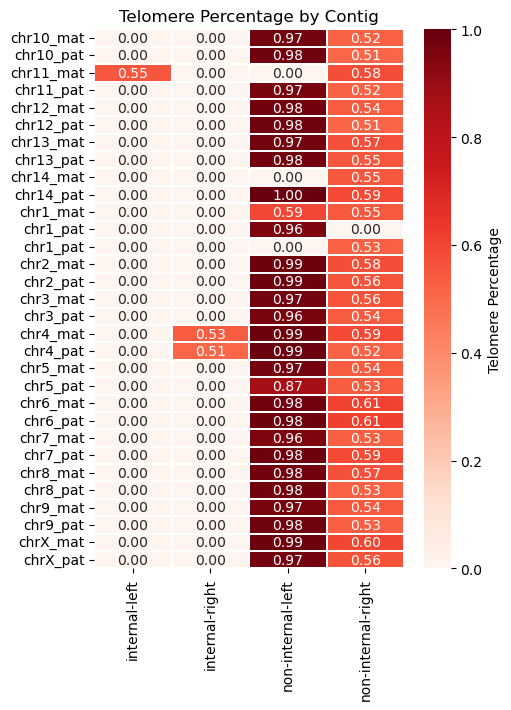
Here, we found a total of three internal telomere sequences from chr11_mat, chr4_mat, and chr4_pat.
However, both haplotypes of chr4 already have a real telomere at the right end of the contig. (Since we don’t yet know which side corresponds to the p or q arm, we refer to them as left and right.)
To further investigate chr11_mat, we will use the vf.pp.find_reads_intra_telo() function to identify which reads were used to the consensus sequence beyond the telomere regions.
vf.pp.find_reads_intra_telo(tel, 2 ,scfmap = "assembly.scfmap",layout = "6-layoutContigs/unitig-popped.layout")
Finding the reads support for the additional artifical sequences outside of the telomere...
Looking for the reads from start of dam_compressed.k31.hapmer-0000002
Looking for the reads from piece000003
Summary :
Num of ONT reads : 6
Num of HiFi reads : 12
| readName | start_hpc | end_hpc | start | end | type | |
|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
We have well-balanced sequencing coverage across the region from the telomere to the internal telomere for chr11_mat, so trimming this region is not necessary.