Final touch to the assembly before polishing#
This Jupyter notebook will guide you on how to create a cleaner assembly after rerunning Verkko to fill some gaps and before proceeding to the polishing step. This includes contig trimming, chromosome flipping, and chromosome renaming.
%load_ext autoreload
%autoreload 2
import sys
import importlib
import pandas as pd
import time
import os
from IPython.display import Image, display
pd.set_option('mode.chained_assignment', None)
from itables import init_notebook_mode
init_notebook_mode(all_interactive=True)
import warnings
import session_info
# Suppress FutureWarnings
warnings.simplefilter(action='ignore', category=FutureWarning)
import verkkofillet as vf
# importlib.reload(vf)
verkkoFillet version 0.1.0
verkkoFillet version 0.1.0
<module 'verkkoFillet' from '/path/to/your/folder/script/post_verkko_pkg/src/verkkoFillet/__init__.py'>
Load data#
verkkoDir="/path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_v0.1.0"
obj = vf.pp.read_Verkko(verkkoDir)
The Verkko fillet target directory already exists: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_v0.1.0_verkko_fillet
If you didn't mean this, please set another directory or for overwirting, please use force= True
Lock the original Verkko folder to prevent it from being modified.
[lock_original_folder] Command executed successfully!
Path file loading...from /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_v0.1.0_verkko_fillet/assembly.paths.tsv
Path file loaded successfully.
scfmap file loading...from /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_v0.1.0_verkko_fillet/assembly.scfmap
scfmap file loaded successfully.
os.chdir(obj.verkko_fillet_dir)
obj
FilletObj
verkkoDir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_v0.1.0
verkko_fillet_dir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_v0.1.0_verkko_fillet
paths: name, path, assignment
history: timestamp, activity
scfmap: contig, pathName
Make stats#
%%time
vf.tl.getT2T(obj)
getT2T was done!
CPU times: user 2.26 ms, sys: 6.25 ms, total: 8.51 ms
Wall time: 6.86 s
ref_fasta = "/path/to/your/folder/ncbiRef/GCA_017591445.1_ASM1759144v1_genomic.rename.fa"
map_file="/path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_legacy/chromosome.map"
%%time
ref = "/path/to/your/folder/ncbiRef/GCA_017591445.1_ASM1759144v1_genomic.rename.fa"
vf.tl.chrAssign(obj = obj, ref = ref)
The [assembly.mashmap.out] file is already exists
If you want to re-run this job, please detete [assembly.mashmap.out]
CPU times: user 217 µs, sys: 87 µs, total: 304 µs
Wall time: 391 µs
obj = vf.pp.readChr(obj, map_file, sire = "pat", dam = "mat")
The chromosome infomation was stored in obj.stats
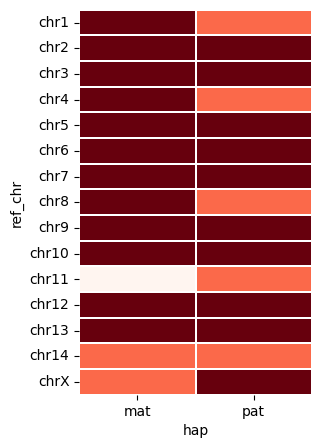
vf.pl.contigPlot(obj)
Trimming#
# vf.tl.run_intra_telo(obj)
file = "assembly.tel_finder.windows.0.5.bed"
vf.pp.find_intra_telo(obj, file=file, loc_from_end=15000)
| contig | old_chr | ref_chr | hap | start | end | len_fai |
|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
trim_contig_dict = { 'contig' : ['dam_compressed.k31.hapmer-0000002'],
'from' : [18800],
'to' : [152521440]
}
vf.tl.runTrimming(obj, trim_contig_dict)
Trim Chromosomes: 0%| | 1/253 [00:00<02:19, 1.80it/s]
dam_compressed.k31.hapmer-0000002 will be trimmed
samtools faidx assembly.fasta dam_compressed.k31.hapmer-0000002:18800-152521440 | sed -e '1d' | sed -e '1i >dam_compressed.k31.hapmer-0000002'>> assembly_trimmed.fasta
Trim Chromosomes: 100%|███████████████████████| 253/253 [00:15<00:00, 16.18it/s]
Trimming completed. Output file: assembly_trimmed.fasta
Flipping chromosome#
If your species already has a reference genome, you can examine the orientation of your assembly. If the orientation is flipped, you can adjust (flip) specific chromosomes as needed. However, it is important to ensure that the previous reference genome was carefully curated for orientation. To confirm this, you can review the methods section in the original publication associated with the released reference genome or other relevant documentation.
output
assembly.mashmap.outassembly.homopolymer-compressed.chr.csvunitigs.hpc.mashmap.outtranslation_hap1translation_hap2chr_completeness_max_hap1chr_completeness_max_hap2
image_path = "/path/to/your/folder/script/post_verkko_pkg/data/test_giraffe/fig/chromosomeFlipping.png"
display(Image(filename=image_path,width=400))
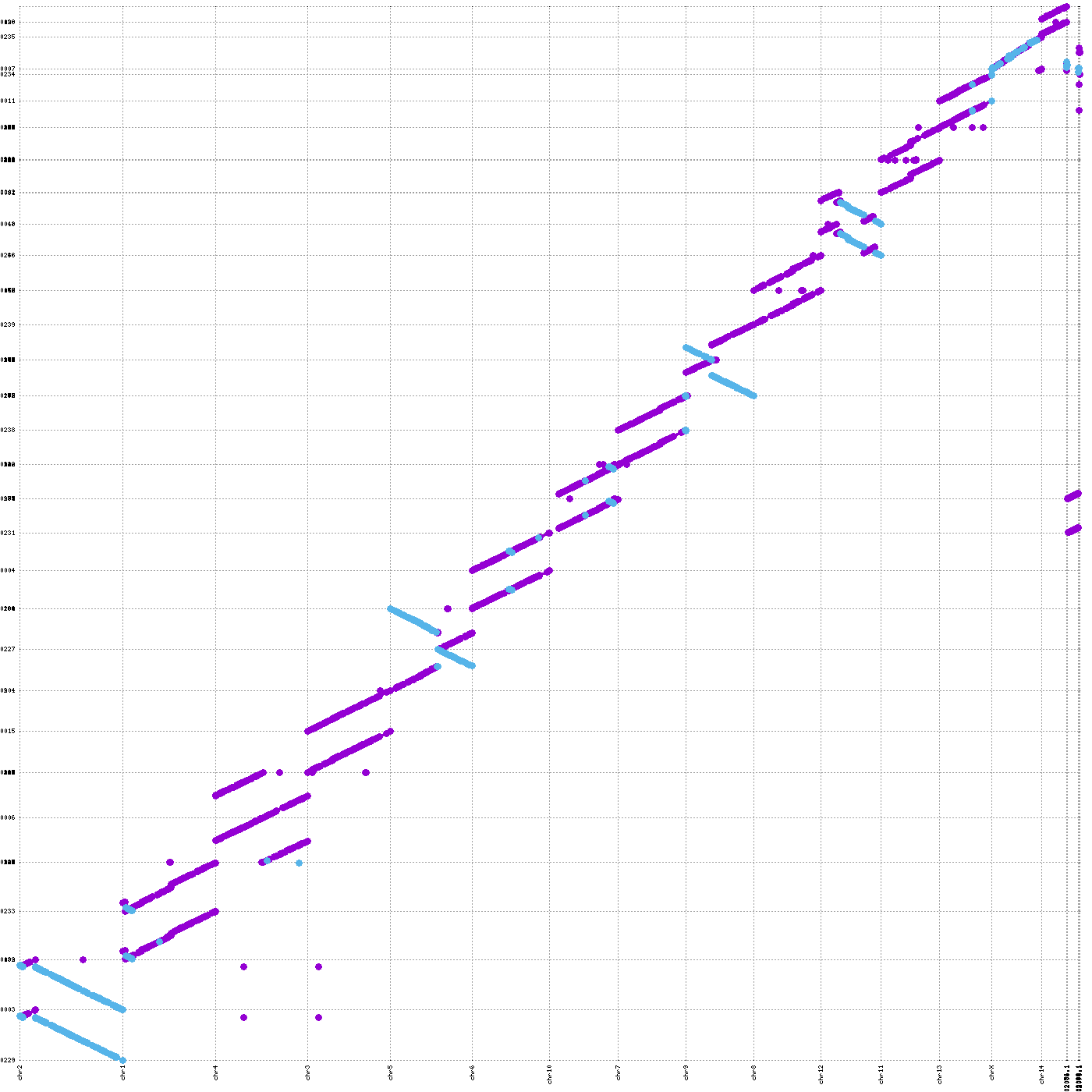
vf.tl.showPairwiseAlign(obj, size="large", minLen = 100000)
[showPairwiseAlign_1] Command executed successfully!
[showPairwiseAlign_2] Command executed successfully!
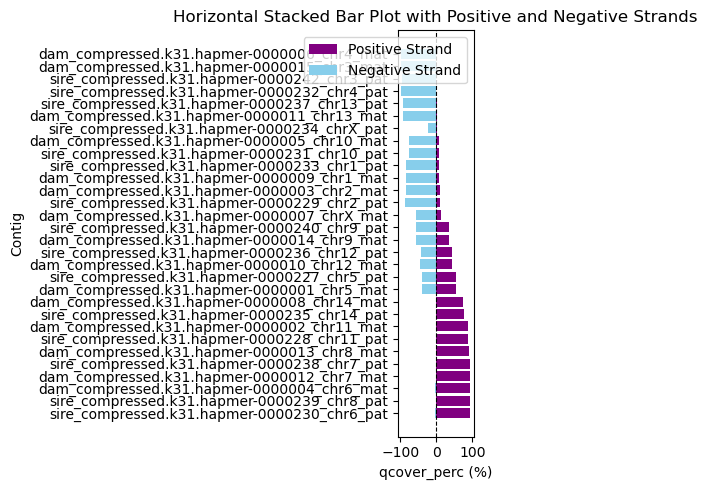
vf.pl.showMashmapOri(obj , by = 'all')
filp_contig_list = ['sire_compressed.k31.hapmer-0000229', #chr2_pat
'dam_compressed.k31.hapmer-0000003', # chr2_mat
'sire_compressed.k31.hapmer-0000242', #chr3_pat
'dam_compressed.k31.hapmer-0000015', #chr3_mat
'sire_compressed.k31.hapmer-0000232', #chr4_mat
'dam_compressed.k31.hapmer-0000006', #chr3_pat
'sire_compressed.k31.hapmer-0000230', #chr6_pat
'dam_compressed.k31.hapmer-0000004', #chr6_mat
'sire_compressed.k31.hapmer-0000238', #chr7_pat
'dam_compressed.k31.hapmer-0000012', #chr7_mat
'sire_compressed.k31.hapmer-0000239', #chr8_pat
'dam_compressed.k31.hapmer-0000013', #chr8_mat
'sire_compressed.k31.hapmer-0000240', #chr9_pat
'dam_compressed.k31.hapmer-0000014',] #chr9_pat
len(filp_contig_list)
14
obj.stats.loc[obj.stats['contig'].isin(filp_contig_list),]
| contig | ref_chr | contig_len | ref_chr_len | hap | old_chr | completeness | hap_verkko | t2tStat | |
|---|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
The vf.tl.flipChr() function flips the original FASTA file using the list of chromosomes, flip_contig_list. The final FASTA file will be generated with the suffix _flipped.fasta.
vf.tl.flipContig(filp_contig_list,
ori_fasta="assembly_trimmed.fasta",
final_fasta="assembly_trimmed_flipped.fasta")
Flipping Chromosomes: 100%|███████████████████| 253/253 [00:58<00:00, 4.31it/s]
The chromosome flipping was completed successfully!
Output FASTA: assembly_trimmed_flipped.fasta
Renaming Primary and Other Small Contigs with Chromosome Information and Haplotype#
We will rename contigs using patterns such as [chr]_[hap] for primary chromosomes and [chr]_[hap]_random_[node] for random contigs, where node represents a representative node in the path. You can trace back the original paths later.
chr: chr1, chr2, chr3, … UnChr
hap: mat, pat, or Unhap
vf.pp.find_multi_used_node() identifies nodes that are used by more than one chromosome. We will exclude these nodes when clustering nodes by chromosome using link information from the GFA file.
You will get two outputs:
duplicate_nodes: A list of nodes used in different chromosomes.node_database: A dataframe with two columns (“chromosome” and “nodeSet”), containing the set of nodes used exclusively in the corresponding chromosome.
image_path = "/path/to/your/folder/script/post_verkko_pkg/data/test_giraffe/fig/nodeClustering.png"
display(Image(filename=image_path,width=400))
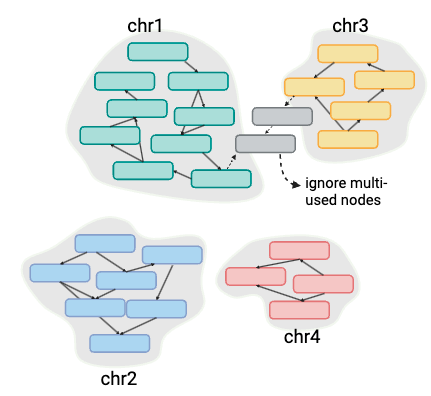
duplicate_nodes, node_database = vf.pp.find_multi_used_node(obj)
# duplicates, node_database = vf.pp.find_multi_used_node(obj)
duplicate_nodes
['utig4-1759', 'utig4-1805', 'utig4-1757']
The vf.pp.naming_contigs() function uses the FAI index of assembly.fasta located in the obj.verkkoDir directory, along with obj.paths and obj.scfmap, to assign the corresponding chromosome and haplotype to non-primary contigs.
chrMap = vf.pp.naming_contigs(obj, node_database, duplicate_nodes)
component_15 : empty
chrMap
| contig | new_contig_name | |
|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
If you have made any changes to the chromosome naming, please edit the chrMap directly before using vf.tl.renameContig().
chrMap['new_contig_name'] = chrMap['new_contig_name'].replace('chrX_pat', 'chrY_pat')
This is the final step. Using the dataframe obtained above, we can rename the contigs using vf.tl.renameContig(). The final FASTA file will be generated with the suffix _rename.fasta.
vf.tl.renameContig(obj, chrMap, original_fasta="assembly_trimmed_flipped.fasta")
[chrRename] Command executed successfully!
Final renamed fasta file : assembly_trimmed_flipped_rename.fasta
Sort#
Before polishing, we will sort the contigs either by chr or by hap:
by
hap: Contigs will be sorted by chromosome within haplotype. For example: chr1_mat > chr2_mat > chr3_mat > … > chrX_mat > chr1_pat > chr2_pat > … > chr1_mat_random_utig4-10 > …by
chr: Contigs will be sorted by haplotype within chromosome. For example: chr1_mat > chr1_pat > chr2_mat > chr2_pat > … > chr1_mat_random_utig4-10 > …
The default mode is sort_by="hap". You can use sort_by="chr", but hap sorting is better when viewing the assembly in IGV.
vf.tl.sortContig("assembly_trimmed_flipped_rename.fasta", sort_by="hap")
Sorted sequences have been written to assembly_trimmed_flipped_rename_sortedhap.fasta