Preprocessing and gap filling#
Verkko-fillet is an easy-to-use toolkit for finishing Verkko assemblies. Here we begin with tools for generating a Verkko-Fillet object, performing assembly quality checks, identifying gaps, assigning chromosomes, searching for ONT reads to help resolve gaps, filling gaps, and generating the final Rukki path (in a GAF-like format) for future Verkko CNS runs.
This Python-based implementation streamlines the entire process. Here we assume the initial Verkko run completed and is ready for the graph curation. The goal is to prepare a post-curated path to walk through the assembly graph for re-doing the final consensus run with minimal manual intervention.
%load_ext autoreload
%autoreload 2
import sys
import importlib
import pandas as pd
import time
import os
from IPython.display import Image, display
pd.set_option('mode.chained_assignment', None)
import matplotlib.pyplot as plt
from itables import init_notebook_mode
init_notebook_mode(all_interactive=True)
import warnings
import session_info
# Suppress FutureWarnings
warnings.simplefilter(action='ignore', category=FutureWarning)
import sys
import tqdm
from importlib import reload
print(os.getcwd())
sys.path.append('/data/Phillippy/projects/giraffeT2T/assembly/script/verkko-fillet/src')
import verkkofillet as vf
vf = reload(vf)
/path/to/your/folder/script/test_notebooks/basics
verkko-fillet version:0.1.21
verkko-fillet version:0.1.21
%load_ext autoreload
%autoreload 2
The autoreload extension is already loaded. To reload it, use:
%reload_ext autoreload
import sys
import importlib
import pandas as pd
import time
import os
from IPython.display import Image, display
pd.set_option('mode.chained_assignment', None)
from itables import init_notebook_mode
init_notebook_mode(all_interactive=True)
import warnings
import session_info
# Suppress FutureWarnings
warnings.simplefilter(action='ignore', category=FutureWarning)
import verkkofillet as vf
importlib.reload(vf)
verkko-fillet version:0.1.21
<module 'verkkofillet' from '/data/Phillippy/projects/giraffeT2T/assembly/script/verkko-fillet/src/verkkofillet/__init__.py'>
The verkkoFillet module, abbreviated as vf, consists of three sub-modules: pp, tl, and pl.
vf.tl: Provides tools for running shell scripts.vf.pp: Includes modules for preprocessing and modifying datasets.vf.pl: Dedicated to plotting and visualization.
Loading verkko directory into the verkko-fillet object#
The input is very simple. You only need the main Verkko output directory. vf.pp.read_Verkko will read all required files, load them into a Verkko-Fillet object, and generate a new folder with the default name {verkkoDir}_verkko-fillet.
verkkoDir = '/mypath/myVerkkoDir'
obj = vf.pp.read_Verkko(verkkoDir)
print(obj.verkko_fillet_dir)
os.chdir(obj.verkko_fillet_dir)
The Verkko fillet target directory already exists: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet
If you didn't mean this, please set another directory or for overwirting, please use force= True
Lock the original Verkko folder to prevent it from being modified.
[lock_original_folder] Command executed successfully!
change working directory: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet
Path file loaded successfully.
scfmap file loaded successfully.
Reading assembly.homopolymer-compressed.noseq.gfa
Reading assembly.colors.csv
Node information is stored in obj.node
/path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet
obj
FilletObj
verkkoDir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic
verkko_fillet_dir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet
paths: name, path, assignment
history: timestamp, activity, function
scfmap: contig, pathName
node: node, len, mat, pat, mat:pat, color, ont_cov, hifi_cov
edge: node1, node1_strand, node2, node2_strand, overlap
The Verkko-fillet object will read all files in the Verkko output directory, including the final path file, ONT alignment, and others, if they exist. In this Jupyter notebook, we will generate all the necessary files and statistics and load them into the Verkko-fillet object later. Once you build your own Verkko Fillet object, you can save it locally as a Python pickle file, allowing you to load it anytime for future curation tasks.
You can use the Verkko-fillet Python module to view and edit the object using specific functions. Additionally, you can easily access each piece of data stored in the object through its attributes (e.g., obj.verkkoDir).
When you print the object, it will display the types of data stored within it.
Stats#
Before starting curation and polishing the assembly, it is helpful to examine the statistics and quality of the initial assembly.
Firstly, run the vf.pp.getT2T function to retrieve the T2T statistics, including the number of scaffolds, contigs, telomeres, and gaps. This function generates four files in the Verkko output directory using the assembly.fasta file:
assembly.telomere.bedassembly.gaps.bedassembly.t2t_scfsassembly.t2t_ctgs
%%time
vf.tl.getT2T(obj)
getT2T was done!
CPU times: user 0 ns, sys: 8.84 ms, total: 8.84 ms
Wall time: 1min 12s
Collapse rDNA nodes.#
As default, vf.tl.rmrDNA function uses the homopolymer-compressed rDNA morphs to screen and collapse rDNA nodes in the Verkko graph, along with the assembly.telomere.bed file generated above using vf.tl.getT2T(obj). This process marks telomere nodes at the end of the contigs and generates three output files:
target.screennodes.out: This file includes the nodes that have rDNA sequences.assembly.homopolymer-compressed.noseq.telo_rdna.gfa: The graph with the rDNA collapsed and telomere nodes added.assembly.colors.telo_rdna.csv: Adds telomere nodes marked in green (#008000) and rDNA nodes marked in purple (#A020F0), in addition to the originalassembly.colors.csv.
%%time
vf.tl.rmrDNA(obj)
Starting removing rDNA nodes in the graph
All output files already exist. Skipping rDNA removal.
CPU times: user 1.17 ms, sys: 19 µs, total: 1.19 ms
Wall time: 2.02 ms
QV calculation#
QV (Quality Value) is a fundamental metric for assessing the quality of an assembly.
The input for QV calculation is a list of high-quality sequencing data, such as HiFi reads, used for the Verkko assembly. K-mers are extracted separately from the sequencing data and the reference assembly. The number of shared k-mers between the two is then used to calculate the QV.
A high QV indicates a high-quality assembly, where most k-mers are shared with the original high-quality sequencing reads.
A low QV suggests that the assembly contains many errors not present in the high-quality sequencing data.
Calculating QV requires a fofn file containing on column of file paths to the fastq files.
fofn = "/path/to/child.fofn"
kmerPrefix="child_illumina"
The k-mer databases for each data and assembly will generated by meryl.
We will use meryl to generate a k-mer database from raw reads using the vf.tl.mkMeryl() function. Then, we will calculate the QV score for each haplotype and the combined assembly by utilizing merqury within the vf.tl.calQV() function.
Please ensure that merqury is installed and update your ~/.profile to include the $MERQURY global variable. This allows it to be used as described in the merqury GitHub repository..
%%time
vf.tl.mkMeryl(obj, fofn, prefix=kmerPrefix)
%%time
vf.tl.calQV(obj, prefix=kmerPrefix)
obj = vf.pp.getQV(obj, f"kmer/{kmerPrefix}.qv_cal.qv")
obj
FilletObj
verkkoDir: /path/to/verkko_result_directory
verkko_fillet_dir: /path/to/verkko_result_directory_verkko_fillet
paths: name, path, assignment
qv: asmName, nKmer_uniq_asm, nKmer_total, QV, ErrorRate
history: timestamp, activity, function
obj.qv
| asmName | nKmer_uniq_asm | nKmer_total | QV | ErrorRate |
|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
Each column shows:
Assembly of interest: This is a combination of the two datasets mentioned above.
K-mers uniquely found only in the assembly
K-mers found in both the assembly and the read set
QV (Quality Value)
Error rate
For more detailed information, please visit the Merqury GitHub.
vf.pl.qvPlot(obj)
/gpfs/gsfs11/users/kimj75/miniforge3/envs/verkko-fillet-local/lib/python3.9/site-packages/verkkofillet/plotting/_baseQC.py:54: UserWarning: set_ticklabels() should only be used with a fixed number of ticks, i.e. after set_ticks() or using a FixedLocator.
ax1.set_xticklabels(ax1.get_xticklabels(), rotation=45, ha='right')
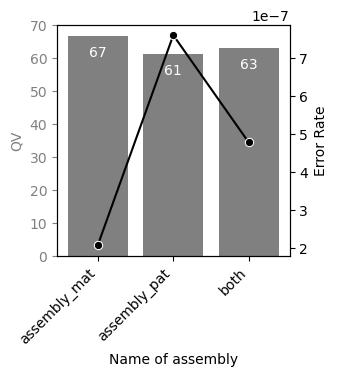
Chromosome assignment#
To check which chromosomes are contigs and to pair the same chromosomes from two haplotypes, chromosome assignment is crucial. The vf.tl.chrAssign function will run Mashmap to align the assembly to a user-provided reference FASTA file and obtain high-confidence chromosome assignments for each contig in the assembly. Once this is done, we will load not only the chromosome assignment results but also the T2T stats generated earlier using the vf.pp.getT2T function.
If you have a reference fasta with chromosome names that are not easy to read, for example start with NC or CM, etc, you can convert the name of the fasta file using vf.tl.convertRefName() with reference fasta and map file.
The chromosome.map file has two columns:
One column contains the names of contigs associated with each primary chromosome (starting with CN, CM, etc., in the reference FASTA file).
The other column contains the names of the primary chromosomes (e.g., chr1, chr2, etc.).
Both columns should be unique. If you have more than one contig from same chromosome, please make them unique(ex. chr1_q, chr1_p)
These contig names need to be converted into easy-to-read chromosome names.
ref_fasta = "/path/to/ref/ncbiRef/GCA_017591445.1_ASM1759144v1_genomic.fna"
map_file= "/path/to/your/folder/ncbiRef/GCA_017591445.1_ASM1759144v1_assembly_chromMap.chr"
# vf.tl.convertRefName(ref_fasta, map_file)
The function vf.tl.convertRefName() generates a FASTA file with the desired names specified in the chromosome.map file. The newly named file will have the suffix .rename.fa.
ref_fasta = "/path/to/ref/ncbiRef/GCA_017591445.1_ASM1759144v1_genomic.rename.fa"
Before running chromosome assignment, we recommend filtering out non-primary reference contigs to avoid assigning chromosome information from alternative or small contigs.
vf.tl.filterContigs function can do that with map_file or list of contigs given to filter_chr_list parameter.
This can be done by running the following command:
%%time
vf.tl.filterContigs(map_file, ref_fasta)
ref_fasta = "/path/to/ref/ncbiRef/GCA_017591445.1_ASM1759144v1_genomic.rename_filtered.fasta"
The vf.tl.chrAssign function uses mashmap to assign chromosomes to each contig in the assembly. If you set force=True, the function will remove all existing outputs in the output folder and restart the process from running mashmap.
vf.tl.chrAssign(obj = obj, ref = ref)
If you have trio or parental information, you can assign which haplotype is paternal and which is maternal using vf.pp.readChr function. If trio information is not available and your contigs are labeled with ‘haplotype’ instead of ‘sire’ or ‘mat,’ these parameters will be ignored.
obj = vf.pp.readChr(obj, map_file)
Haplotype names not provided, using default haplotype identifiers.
The chromosome infomation was stored in obj.stats
obj
FilletObj
verkkoDir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic
verkko_fillet_dir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet
paths: name, path, assignment
stats: contig, ref_chr, contig_len, ref_chr_len, hap, old_chr, completeness, hap_verkko, t2tStat
history: timestamp, activity, function
scfmap: contig, pathName
node: node, len, mat, pat, mat:pat, color, ont_cov, hifi_cov
edge: node1, node1_strand, node2, node2_strand, overlap
obj.stats.head(2)
| contig | ref_chr | contig_len | ref_chr_len | hap | old_chr | completeness | hap_verkko | t2tStat |
|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
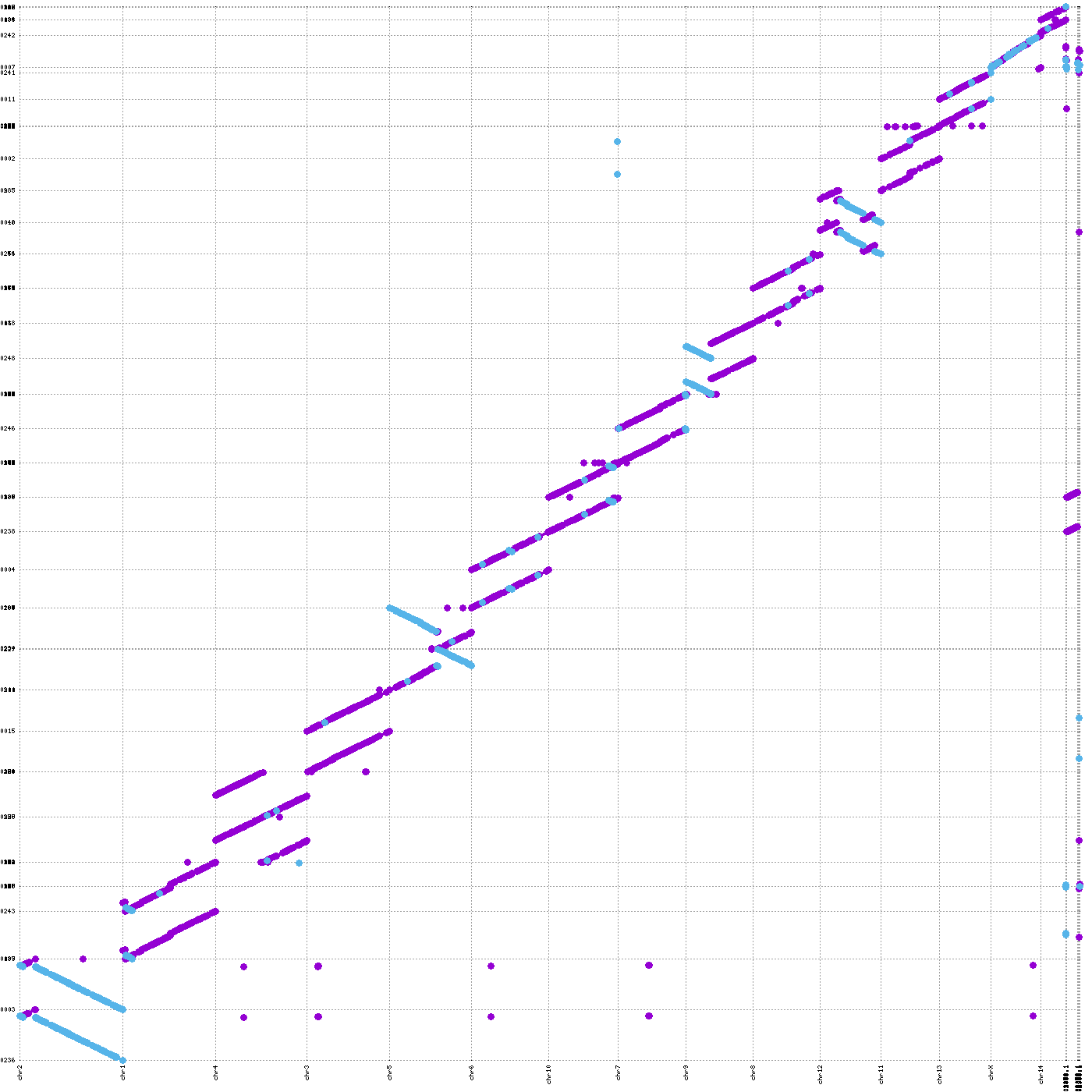
To identify orientation discrepancies or structural differences between the assembly and the reference, we provide the showPairwiseAlign function. This function aligns the assembly to the reference and visualizes the resulting PAF file as a dot plot. The colors indicate the alignment strand: blue represents reverse-strand alignments, and purple represents forward-strand alignments.
If a contig shows a completely negative alignment, the contig should be flipped relative to the reference. If mixed orientations are observed, as in the chr5 example, this indicates potential structural discrepancies between the assembly and the reference, which should be further inspected in both datasets.
vf.tl.showPairwiseAlign(obj, size="large")
[showPairwiseAlign_1] Command executed successfully!
[showPairwiseAlign_2] Command executed successfully!
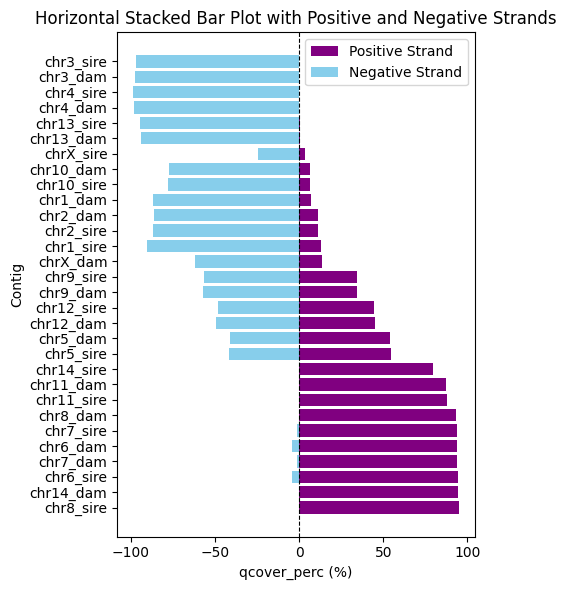
Another way to identify orientation discrepancies is to generate a stacked bar plot showing the total length of alignment blocks between the assembly and the reference. As mentioned earlier, if a large proportion of the alignment blocks are on the negative strand, this indicates that the contig should be flipped. If both strand orientations are present, it suggests a structural discrepancy between the assembly and the reference.
vf.pl.showMashmapOri(obj, height = 6, width = 5)
File figs/intra_telo.heatmap.png already exists
Please remove the file or change the name
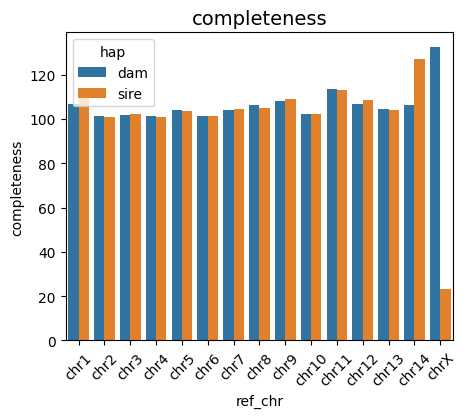
Completeness is the ratio of the assembled contig length to the length of the matched reference contig, indicating how many bases were assembled relative to the reference. A value of 100 indicates that the assembled and reference contigs have the same length, while a value greater than 100 indicates that the assembled contig is longer than the reference contig.
vf.pl.completePlot(obj, height = 4 , width = 5)
File figs/completePlot.barplot.png already exists
Please remove the file or change the name
vf.pl.contigLenPlot(obj,height = 5 , width = 10)
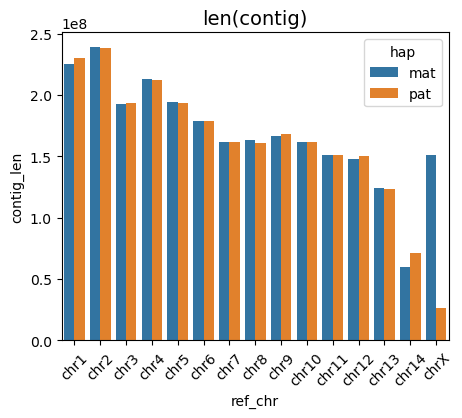
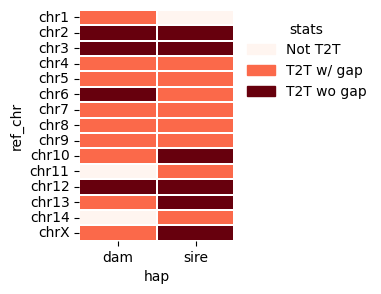
We can also visualize the T2T status of each contig using files generated by the getT2T() function. This brick plot displays the T2T status, with colors indicating different assembly categories: dark brick represents gap-free T2T contigs, salmon represents T2T contigs with one or more gaps, and beige represents contigs missing one or both telomeres.
red brown(#5E0E12): Contig (complete T2T without gaps)
Salmon Color(#E97254): Scaffold (T2T with gaps or tangles)
Beige(#FDF5F1) : Not a scaffold (missing one of the telomere)
vf.pl.contigPlot(obj,height = 3 , width = 2)
File figs/contigPlot.heatmap.png already exists
Please remove the file or change the name
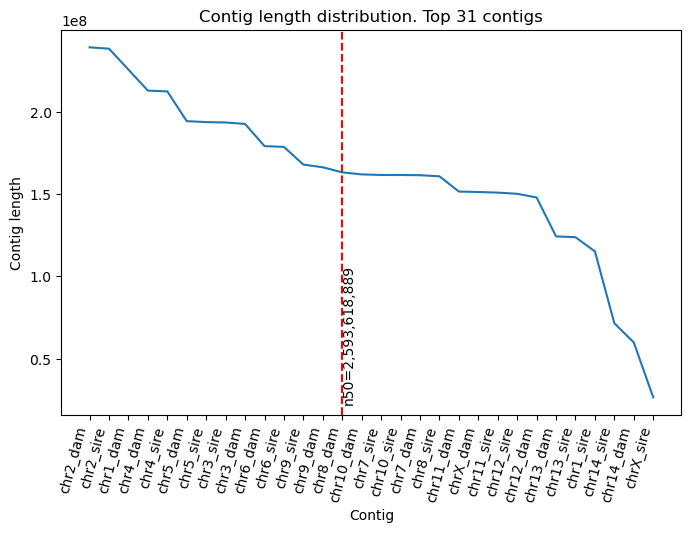
vf.pl.n50Plot(obj)
File figs/N50.png already exists
Please remove the file or change the name to save the new file
n50=2,593,618,889
l50=13
Lets fillet the verkko assembly!#
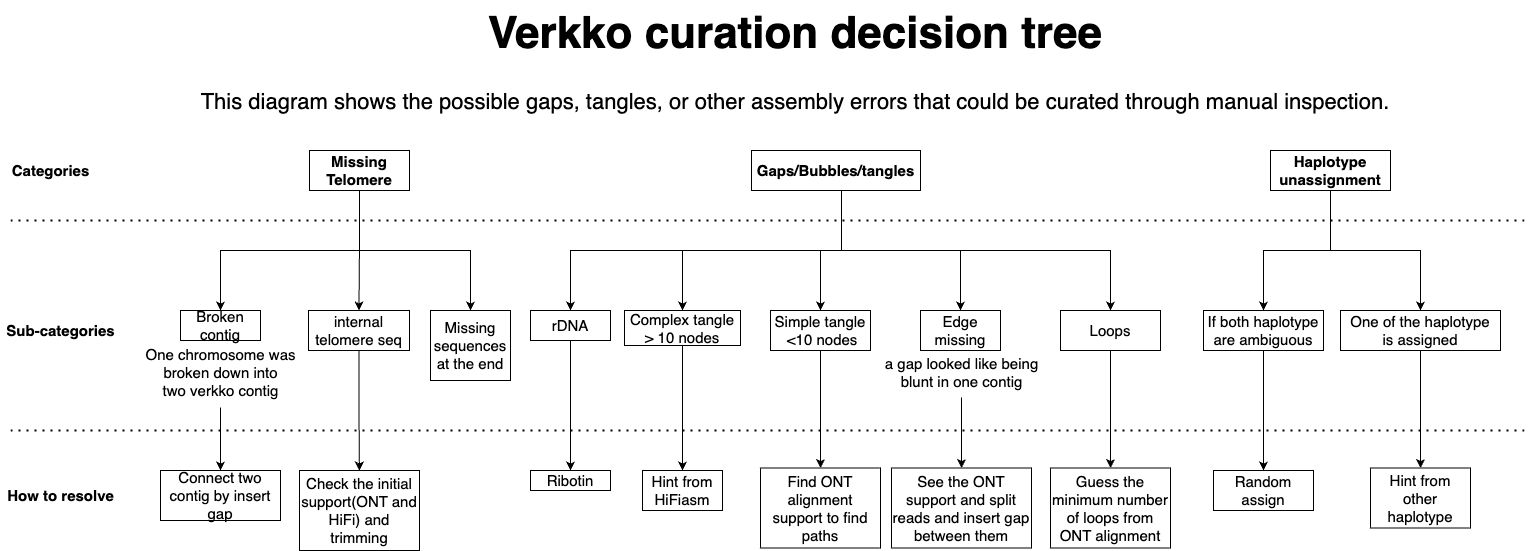
Verkko is a very powerful tool for assembling long-read data to generate a complete diploid assembly, but it still has some limitations, such as generating gaps, tangles, missing telomeres, and more. These issues can occur due to sequencing errors, lower sample quality, or overly complex or homogeneous sequences, which are difficult to assemble. As a result, some manual inspection is needed to identify errors and correct them (“curation”). Below, we classify the scenarios that can lead to gaps in the assembly and their potential solutions.
1. Missing Telomeres
Broken Contig: This can occur when large tangles or gaps exist in the middle of a contig, causing Rukki to split the contig. This can be resolved by connecting the contigs with a gap in the Rukki path.
Internal Telomere Sequencing: In this case, additional sequences are added after the telomere, preventing the
getT2Tfunction from detecting the telomere and reporting the contig as a scaffold, even if it contains an internal telomere. This issue might arise due to ONT sequencing bias and can be fixed by trimming the sequences before the internal telomere after identifying its start position.Missing Sequence at the End: If you see isolated telomere-attached nodes, you can find homologous nodes to determine the counterpart of the haplotype on the chromosome and stitch them together by inserting a gap.
2. Gaps, Bubbles, and Tangles
rDNA: The number of rDNA arrays and their morphs vary by species, and rDNA sequences are too similar to be assembled automatically. We recommend running
ribotinto find the consensus of the rDNA sequences and patch the assembly with the rDNA consensus.Complex Tangles: Sometimes the genome assembler Hifiasm can generate longer contigs that cover tangles in the Verkko assembly. We can align the HiFI assembly to the Verkko graph to reveal hints how to resolve the walk based on node order and orientation.
Simple Tangles or Bubbles: In this scenario, ONT alignments on the Verkko graph using
graphalignercan be helpful. Searching for supported paths that connect nodes in the gap can help resolve simple tangles.Edge Missing: If your assembly has missing edges between two nodes, this might be due to too few supporting ONT reads (<3) to connect the nodes. We can find the supported split ONT reads aligned on nearby nodes and add the edge to the graph.
Loops: Repeated sequences with multiple copies can be problematic to assemble, especially when the repeat is long enough to span ONT reads at both ends of the flanking nodes. In this case, we edit the gaps with the estimated number of loops and fill the gaps with semi-filled gaps.
3. Unassigned Haplotypes
One Haplotype is Ambiguous: This scenario occurs when heterozygous nodes flank long runs of homozygous nodes, making Verkko unsure which haplotype should be assigned. If we have evidence that the other haplotype has already been assigned to one of the nodes in the gap, we can assign the other haplotype to the ambiguous node.
Both Haplotypes are Ambiguous: When both haplotypes have gaps in a bubble, there’s no way to determine the correct haplotype for the gap. In this case, we will randomly assign the haplotypes. This can be corrected during the polishing step later.
image_path = os.path.dirname(vf.__file__) + "/data/test_giraffe/fig/verkko_curation_decision_tree.png"
display(Image(filename=image_path,width=1500, height = 500))
Missing Telomere#
Please see the Recovering Telomere-to-Telomere Contigs section to check for any internal telomeric sequences. You may be able to recover the T2T contig by trimming noisy, misplaced, or erroneous chimeric ONT reads at the ends of the non-T2T contig.
Gaps / Bubbles / Tangles#
Finding gaps from path#
Using the Rukki final paths that are already loaded in the object, we can retrieve the gaps with flanking nodes and the names of the contigs using the vf.pp.findGaps function. This function will store the list of gaps in the obj.gaps attribute, and you can access the table.
obj
FilletObj
verkkoDir: /path/to/verkko_result_directory
verkko_fillet_dir: /path/to/verkko_result_directory_verkko_fillet
paths: name, path, assignment
stats: contig, ref_chr, contig_len, ref_chr_len, hap, old_chr, completeness, hap_verkko, t2tStat
qv: asmName, nKmer_uniq_asm, nKmer_total, QV, ErrorRate
history: timestamp, activity, function
%%time
obj = vf.pp.findGaps(obj)
45 gaps were found -> obj.gaps
CPU times: user 112 ms, sys: 1.76 ms, total: 114 ms
Wall time: 114 ms
print(obj)
FilletObj
verkkoDir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic
verkko_fillet_dir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet
paths: name, path, assignment, gaps
gaps: gapId, name, gaps, notes, fixedPath, startMatch, endMatch, finalGaf, done
gaf: qname, qlen, qstart, qend, strand, pname, plen, pstart, pend, nmatch, blocksize, mapq, nm, as, dv, id, path_modi
paths_freq: pname, nsupport, path_modi
history: timestamp, activity, function
scfmap: contig, pathName
The first three columns are generated from the Rukki path, and the others are the columns we will fill during gap filling. The gaps column contains each gap along with the flanking nodes, allowing you to easily visualize how the gaps or bubbles look using the flanking nodes in Bandage.
obj.gaps
| gapId | name | gaps | notes | fixedPath | startMatch | endMatch | finalGaf | done | |
|---|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
Align ONT Reads onto the graph#
All ONT datasets used during the Verkko assembly are stored in the Verkko output directory. We use the ONT reads and align them to the Verkko graph using graphAligner. This alignment helps us create reasonable paths through the gaps and bubbles. In future versions of Verkko, this step will be integrated into the assembly process.
vf.tl.graphIdx() will index the assembly.homopolymer-compressed.gfa graph, and vf.tl.graphAlign() uses the ONT reads under the 3-align/split directory. The output GAF file will be saved to the 9-graphAlignment/verkko.graphAlign_allONT.gaf.
vf.tl.graphIdx(obj,threads=1)
ont.list is a file containing a single column of ont.fastq or ont.fasta filenames. By default, this is set to None. If not provided, the script will search for the ONT file list used during the Verkko assembly, located under the 3-align/split/ directory in the original Verkko directory.
vf.tl.graphAlign(obj = obj, threads=50, ontReadList = "ont.list")
obj = vf.pp.readGaf(obj)
Looking for GAF file at: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet/graphAlignment/verkko.graphAlign_allONT.gaf
Loading ONT alignment GAF file...
GAF file successfully loaded.
obj
FilletObj
verkkoDir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic
verkko_fillet_dir: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet
paths: name, path, assignment, gaps
stats: contig, ref_chr, contig_len, ref_chr_len, hap, old_chr, completeness, hap_verkko, t2tStat
gaps: gapId, name, gaps, notes, fixedPath, startMatch, endMatch, finalGaf, done
gaf: qname, qlen, qstart, qend, strand, pname, plen, pstart, pend, nmatch, blocksize, mapq, nm, as, dv, id, path_modi
history: timestamp, activity, function
scfmap: contig, pathName
node: node, len, mat, pat, mat:pat, color, ont_cov, hifi_cov
edge: node1, node1_strand, node2, node2_strand, overlap
obj.gaf.head(2)
| Qname | len | path | mapQ | identity | path_modi |
|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
Searching the nodes in the ONT alignment GAF file.#
Once you have loaded the ONT alignment into your Verkko Fillet object, you can search using two nodes to view the number of paths that span both nodes using the vf.pp.searchNodes function. The output will have 4 columns:
path: The ONT alignment path.
size: The number of supported reads for the path.
node1:
Yif the path spans node1.node2:
Yif the path spans node2.
%%time
node_list_input = ['utig4-317', 'utig4-2181']
vf.pp.searchNodes(obj, node_list_input)
`obj.paths_freq` already exists.
skip generating `obj.paths_freq`.
Extracting paths containing nodes: ['utig4-317', 'utig4-2181']
CPU times: user 10.3 ms, sys: 1.41 ms, total: 11.7 ms
Wall time: 11.1 ms
| path | utig4-317 | utig4-2181 | fw | rv | total_support |
|---|---|---|---|---|---|
| >utig4-317<utig4-2324<utig4-2181 | Y | Y | 2 | 0 | 2 |
| <utig4-317 | Y | 10158 | 10093 | 20251 | |
| <utig4-2181 | Y | 421 | 391 | 812 | |
| <utig4-317>utig4-316 | Y | 90 | 86 | 176 | |
| <utig4-2324<utig4-2181 | Y | 88 | 84 | 172 | |
| >utig4-2324<utig4-317 | Y | 85 | 76 | 161 | |
| <utig4-2181>utig4-2180 | Y | 88 | 0 | 88 | |
| <utig4-2180>utig4-2181 | Y | 84 | 0 | 84 | |
| <utig4-317>utig4-316>utig4-319 | Y | 1 | 1 | 2 | |
| <utig4-2181>utig4-2180<utig4-1681 | Y | 1 | 0 | 1 | |
| <utig4-317>utig4-316>utig4-320 | Y | 1 | 0 | 1 |
%%time
node_list_input = ['utig4-424', 'utig4-1283']
vf.pp.searchNodes(obj, node_list_input)
`obj.paths_freq` already exists.
skip generating `obj.paths_freq`.
Extracting paths containing nodes: ['utig4-424', 'utig4-1283']
CPU times: user 9.94 ms, sys: 1.67 ms, total: 11.6 ms
Wall time: 11.1 ms
| path | utig4-424 | utig4-1283 | fw | rv | total_support |
|---|---|---|---|---|---|
| <utig4-1283<utig4-1281<utig4-424<utig4-421 | Y | Y | 1 | 0 | 1 |
| <utig4-1283 | Y | 46356 | 46051 | 92407 | |
| <utig4-1283<utig4-1281 | Y | 80 | 70 | 150 | |
| >utig4-421>utig4-424>utig4-1281 | Y | 66 | 61 | 127 | |
| <utig4-2456<utig4-1283 | Y | 89 | 0 | 89 | |
| >utig4-1283>utig4-2456 | Y | 73 | 0 | 73 | |
| <utig4-424<utig4-421 | Y | 7 | 2 | 9 | |
| <utig4-1281<utig4-424 | Y | 1 | 1 | 2 | |
| >utig4-1901<utig4-2456<utig4-1283 | Y | 1 | 0 | 1 |
vf.pp.highlight_nodes(obj, node = "utig4-1282+")
| name | path |
|---|---|
| dam_compressed.k31.hapmer_from_utig4-1282 | utig4-1774-,utig4-1771-,utig4-1773+,utig4-2684+,utig4-2600-,utig4-2596-,utig4-2597+,utig4-2636-,utig4-1227-,utig4-1225-,utig4-878-,utig4-875-,utig4-877+,utig4-2373+,utig4-2374+,utig4-2452-,utig4-692-,utig4-688+,utig4-693+,utig4-1779+,utig4-1781+,utig4-2402+,utig4-1978-,utig4-1974-,utig4-1976+,utig4-2502+,utig4-2018-,utig4-2017+,utig4-2020+,utig4-2676-,utig4-223-,utig4-221+,utig4-225+,utig4-1809+,utig4-1810+,utig4-2721+,utig4-1790-,utig4-1786-,utig4-1787+,utig4-1993-,utig4-1995+,utig4-2426+,utig4-2427+,utig4-2575-,utig4-721-,utig4-720-,utig4-308-,utig4-306+,utig4-309+,utig4-983+,utig4-984+,utig4-2448+,utig4-415-,utig4-413-,utig4-398-,utig4-394-,utig4-395+,utig4-2602-,utig4-1857-,utig4-1855+,utig4-949-,utig4-946-,utig4-947+,utig4-2268+,utig4-2269+,utig4-2642+,utig4-515-,utig4-512-,utig4-513+,utig4-2161+,utig4-1793-,utig4-1791+,utig4-1470-,utig4-1469-,utig4-1251-,utig4-1250+,utig4-1253+,utig4-2573-,utig4-199-,utig4-197+,utig4-200+,utig4-2000+,utig4-2002+,utig4-2403+,utig4-1642-,utig4-1592-,utig4-1589+,utig4-1593+,utig4-1818-,utig4-1819+,utig4-2388+,utig4-2390+,utig4-2490-,utig4-2386-,utig4-2384+,utig4-2257-,utig4-2255-,utig4-474-,utig4-472+,utig4-475+,utig4-2363+,utig4-2327-,utig4-2325+,utig4-2328+,utig4-2650-,utig4-422-,utig4-421+,utig4-425+,utig4-1281+,utig4-1282+,utig4-2456+,utig4-1902-,utig4-1898-,utig4-1899+,utig4-2106-,utig4-2108+,utig4-2305+,utig4-2192-,utig4-2189-,utig4-2190+,utig4-2238+,utig4-1077-,utig4-1074-,utig4-1076+,utig4-2166-,utig4-1101-,utig4-1098-,utig4-1100+,utig4-2751-,utig4-2752+ |
In this scenario, we find one supporting ONT alignment spanning the gap and flanking reads. However, the number of supporting reads is too small, so we also check the other haplotype paths by using the vf.pp.highlight_nodes() function with the counterpart nodes on the other haplotype to see if another haplotype uses the other nodes in the bubble. If so, as shown here, we can assign the node utig-424 to the paternal strand to fill the gap.
If you have enough evidence for filling the gaps (or partially filling them), you can use this information to fill the gaps using the vf.pp.fillGaps function. You can easily extract the order and direction of each node using Bandage tools (Output > Specify exact path for copy/save). This information will be stored in the obj.gaps attribute and will be used for generating the edited Rukki path for the Verkko CNS run.
Caution: Be careful to maintain the original node direction when filling the gaps.
%%time
obj = vf.pp.fillGaps(obj=obj,
gapId='gapid_26',
final_path="utig4-424+, utig4-1281+, utig4-1283+")
final path : >utig4-424>utig4-1281>utig4-1283
Updated gapId gapid_26!
✅ The start node and its direction match the original node.
✅ The start node and its direction match the original node.
[== ] 2/45 gaps filled
CPU times: user 2.39 ms, sys: 33 µs, total: 2.42 ms
Wall time: 2.43 ms
obj.history
| timestamp | activity | function |
|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
Once you edit your path, it would be great to check the obj.gaps to ensure that both the startMatch and the endMatch are set to “match.” The vf.pp.fillGaps() function also provides this information when you edit the path. These two pieces of information not only indicate the starting and ending nodes but also account for the direction of the nodes. If you provide reverted nodes, each field will be filled with “notMatch.” If this is unintentional, it should be corrected before completing the gap-filling process.
Here, we show how the vf.pp.fillGaps() function reports when we provide wrong directions for the end nodes.
%%time
obj = vf.pp.fillGaps(obj=obj,
gapId='gapid_26',
final_path="utig4-424+, utig4-1281+, utig4-1283-")
final path : >utig4-424>utig4-1281<utig4-1283
Updated gapId gapid_26!
✅ The start node and its direction match the original node.
❌ The start node and its direction do not match the original node.
[== ] 2/45 gaps filled
CPU times: user 2.4 ms, sys: 0 ns, total: 2.4 ms
Wall time: 2.41 ms
It says that the start node and its direction match, but the end node does not match.
If you want to modify the gap information you have already added, simply use the same function to overwrite it. Additionally, if you want to reset the gap information, set final_path="" in vf.pp.fillGaps.
%%time
obj = vf.pp.fillGaps(obj=obj,
gapId='gapid_26',
final_path="utig4-424+, utig4-1281+, utig4-1283+")
final path : >utig4-424>utig4-1281>utig4-1283
Updated gapId gapid_26!
✅ The start node and its direction match the original node.
✅ The start node and its direction match the original node.
[== ] 2/45 gaps filled
CPU times: user 2.4 ms, sys: 0 ns, total: 2.4 ms
Wall time: 2.41 ms
Edge Missing#
image_path = os.path.dirname(vf.__file__) + "/data/test_giraffe/fig/unit17.png"
display(Image(filename=image_path,width=500, height = 500))
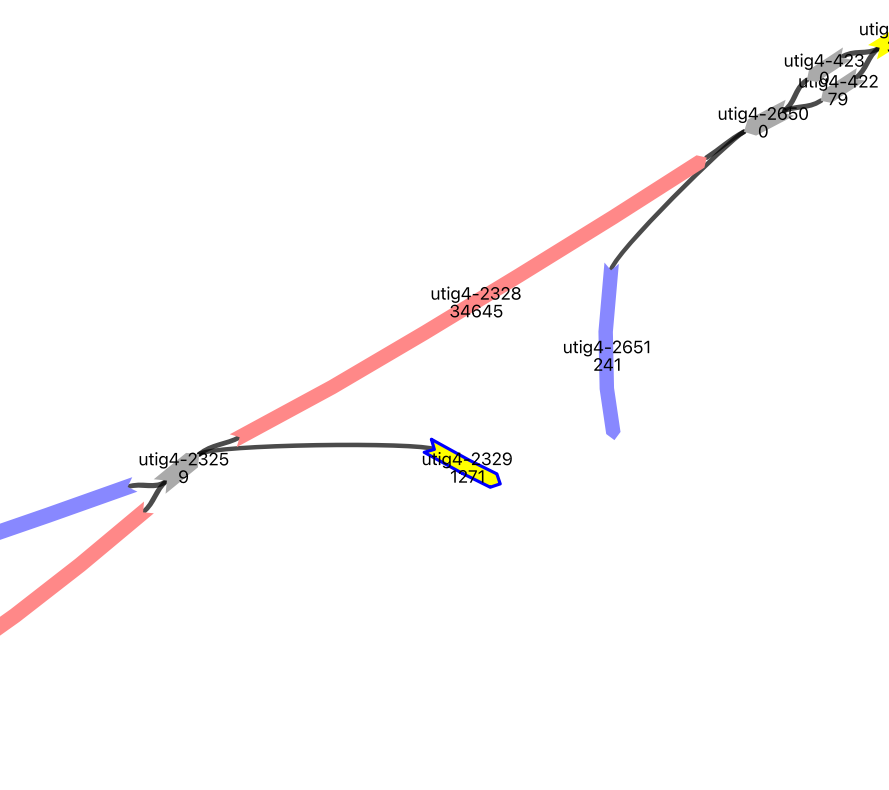
The vf.pp.searchSplit function will find the split reads that are aligned to two nodes that are not connected.
%%time
node_list_input = ["utig4-2329","utig4-2651"]
splitreads = vf.pp.searchSplit(obj,node_list_input)
splitreads
2 reads were found that contain both nodes ['utig4-2329', 'utig4-2651']
CPU times: user 4.77 s, sys: 870 ms, total: 5.64 s
Wall time: 5.67 s
| qname | path_modi |
|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
Verkko connects two nodes only if they have enough, at least 3 ONT reads that can walk on them. However, here we find two nodes such that Verkko cannot create a bridge between them. We can create a new node between the two nodes using the split read dataframe.
%%time
vf.tl.insertGap(obj, gapid="gapid_25", split_reads=splitreads)
Extracting reads...
The split reads for gapid_25 were saved to /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_verkko_fillet/missing_edge/gapid_25.missing_edge.ont_list.txt
The gap filling was completed for gapid_25!
The final path looks like:
{'>utig4-2329>gapmanual-1-len--4492-cov-1<utig4-2651'}
CPU times: user 105 µs, sys: 195 ms, total: 195 ms
Wall time: 19.7 s
%%time
obj = vf.pp.fillGaps(obj=obj,
gapId='gapid_25',
final_path="utig4-2329+,gapmanual-1-len--4492-cov-1+,utig4-2651-")
final path : >utig4-2329>gapmanual-1-len--4492-cov-1<utig4-2651
Updated gapId gapid_25!
✅ The start node and its direction match the original node.
✅ The start node and its direction match the original node.
[====== ] 6/45 gaps filled
CPU times: user 868 ms, sys: 829 ms, total: 1.7 s
Wall time: 1.7 s
Loops#
image_path = os.path.dirname(vf.__file__) + "/data/test_giraffe/fig/unit2.png"
display(Image(filename=image_path,width=500, height = 500))
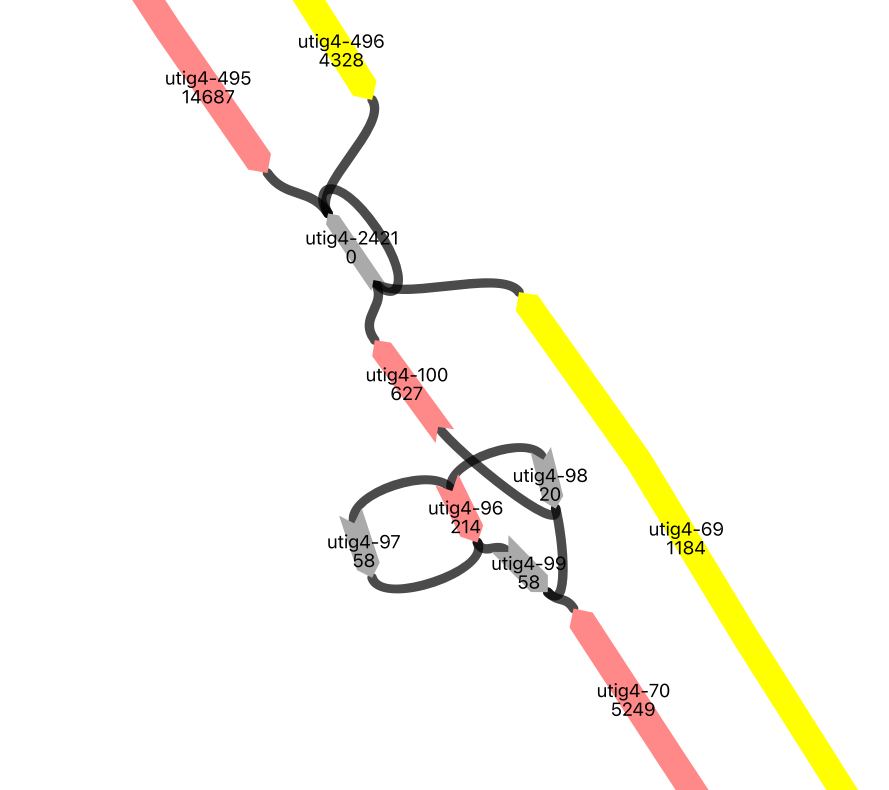
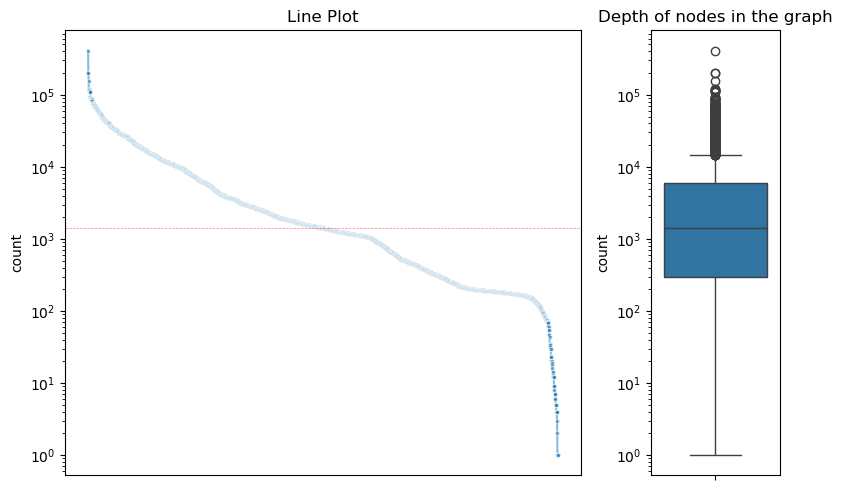
obj = vf.pp.calNodeDepth(obj)
Counting the frequency of each node in the paths...
Median normalized by length: 0.007214537059447785
Median of haplotype specific nodes normalized by length : 0.005721821461611718
Plotting the depth of nodes in the graph...
File figs/nodeDepth.png already exists
Please remove the file or change the name
node = "utig4-100, utig4-2421, utig4-495, utig4-496, utig4-69".split(", ")
obj.node.loc[obj.node['node'].isin(node),:]
| node | len | mat | pat | mat:pat | color | hap | count | norm_len | cov | cov_hap | |
|---|---|---|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
%%time
node_list_input = ['utig4-2421', 'utig4-100']
vf.pp.searchNodes(obj, node_list_input)
`obj.paths_freq` already exists.
skip generating `obj.paths_freq`.
Extracting paths containing nodes: ['utig4-2421', 'utig4-100']
CPU times: user 11.7 ms, sys: 93 µs, total: 11.8 ms
Wall time: 11.2 ms
| path | utig4-2421 | utig4-100 | fw | rv | total_support |
|---|---|---|---|---|---|
| >utig4-495<utig4-2421<utig4-2421<utig4-2421<utig4-100 | Y | Y | 18 | 8 | 26 |
| >utig4-100>utig4-2421 | Y | Y | 11 | 10 | 21 |
| >utig4-100>utig4-2421<utig4-496 | Y | Y | 5 | 4 | 9 |
| >utig4-100 | Y | 906 | 808 | 1714 | |
| <utig4-96>utig4-98>utig4-100 | Y | 37 | 23 | 60 | |
| <utig4-2421 | Y | 16 | 16 | 32 | |
| >utig4-496<utig4-2421 | Y | 17 | 14 | 31 | |
| <utig4-2421<utig4-69 | Y | 13 | 10 | 23 | |
| >utig4-2421<utig4-495 | Y | 8 | 7 | 15 | |
| >utig4-69>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421<utig4-496 | Y | 5 | 3 | 8 | |
| >utig4-69>utig4-2421>utig4-2421<utig4-495 | Y | 3 | 2 | 5 | |
| >utig4-495<utig4-2421<utig4-69 | Y | 1 | 0 | 1 |
%%time
node_list_input = ['utig4-2421', 'utig4-495']
vf.pp.searchNodes(obj, node_list_input)
`obj.paths_freq` already exists.
skip generating `obj.paths_freq`.
Extracting paths containing nodes: ['utig4-2421', 'utig4-495']
CPU times: user 10.8 ms, sys: 978 µs, total: 11.8 ms
Wall time: 11.1 ms
| path | utig4-2421 | utig4-495 | fw | rv | total_support |
|---|---|---|---|---|---|
| >utig4-495<utig4-2421<utig4-2421<utig4-2421<utig4-100 | Y | Y | 18 | 8 | 26 |
| >utig4-2421<utig4-495 | Y | Y | 8 | 7 | 15 |
| >utig4-69>utig4-2421>utig4-2421<utig4-495 | Y | Y | 3 | 2 | 5 |
| >utig4-495<utig4-2421<utig4-69 | Y | Y | 1 | 0 | 1 |
| <utig4-495 | Y | 19027 | 18224 | 37251 | |
| <utig4-494>utig4-495 | Y | 89 | 85 | 174 | |
| <utig4-2421 | Y | 16 | 16 | 32 | |
| >utig4-496<utig4-2421 | Y | 17 | 14 | 31 | |
| <utig4-2421<utig4-69 | Y | 13 | 10 | 23 | |
| >utig4-100>utig4-2421 | Y | 11 | 10 | 21 | |
| >utig4-100>utig4-2421<utig4-496 | Y | 5 | 4 | 9 | |
| >utig4-69>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421<utig4-496 | Y | 5 | 3 | 8 |
%%time
node_list_input = ['utig4-2421', 'utig4-496']
vf.pp.searchNodes(obj, node_list_input)
`obj.paths_freq` already exists.
skip generating `obj.paths_freq`.
Extracting paths containing nodes: ['utig4-2421', 'utig4-496']
CPU times: user 11.6 ms, sys: 72 µs, total: 11.6 ms
Wall time: 11.1 ms
| path | utig4-2421 | utig4-496 | fw | rv | total_support |
|---|---|---|---|---|---|
| >utig4-496<utig4-2421 | Y | Y | 17 | 14 | 31 |
| >utig4-100>utig4-2421<utig4-496 | Y | Y | 5 | 4 | 9 |
| >utig4-69>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421<utig4-496 | Y | Y | 5 | 3 | 8 |
| <utig4-496 | Y | 17143 | 16587 | 33730 | |
| <utig4-496>utig4-494 | Y | 94 | 83 | 177 | |
| <utig4-2421 | Y | 16 | 16 | 32 | |
| >utig4-495<utig4-2421<utig4-2421<utig4-2421<utig4-100 | Y | 18 | 8 | 26 | |
| <utig4-2421<utig4-69 | Y | 13 | 10 | 23 | |
| >utig4-100>utig4-2421 | Y | 11 | 10 | 21 | |
| >utig4-2421<utig4-495 | Y | 8 | 7 | 15 | |
| >utig4-69>utig4-2421>utig4-2421<utig4-495 | Y | 3 | 2 | 5 | |
| >utig4-495<utig4-2421<utig4-69 | Y | 1 | 0 | 1 |
%%time
node_list_input = ['utig4-2421', 'utig4-69']
vf.pp.searchNodes(obj, node_list_input)
`obj.paths_freq` already exists.
skip generating `obj.paths_freq`.
Extracting paths containing nodes: ['utig4-2421', 'utig4-69']
CPU times: user 12.8 ms, sys: 118 µs, total: 12.9 ms
Wall time: 12.3 ms
| path | utig4-2421 | utig4-69 | fw | rv | total_support |
|---|---|---|---|---|---|
| <utig4-2421<utig4-69 | Y | Y | 13 | 10 | 23 |
| >utig4-69>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421<utig4-496 | Y | Y | 5 | 3 | 8 |
| >utig4-69>utig4-2421>utig4-2421<utig4-495 | Y | Y | 3 | 2 | 5 |
| >utig4-495<utig4-2421<utig4-69 | Y | Y | 1 | 0 | 1 |
| >utig4-69 | Y | 6285 | 6153 | 12438 | |
| <utig4-69<utig4-66 | Y | 76 | 0 | 76 | |
| >utig4-66>utig4-69 | Y | 62 | 0 | 62 | |
| >utig4-2421 | Y | 16 | 16 | 32 | |
| >utig4-496<utig4-2421 | Y | 17 | 14 | 31 | |
| >utig4-495<utig4-2421<utig4-2421<utig4-2421<utig4-100 | Y | 18 | 8 | 26 | |
| >utig4-100>utig4-2421 | Y | 11 | 10 | 21 | |
| >utig4-2421<utig4-495 | Y | 8 | 7 | 15 | |
| >utig4-100>utig4-2421<utig4-496 | Y | 5 | 4 | 9 | |
| <utig4-68>utig4-66>utig4-69 | Y | 1 | 0 | 1 |
This is one of the rDNA clusters in the Giraffe genome. The node utig4-2421 has a loop, and we don’t know how many times the node exists in each haplotype strand. Here, we estimate the minimum number of loops for the node on the maternal strand by finding the number of loop nodes in each ONT alignment that spans the haplotype-assigned nodes near the loop node(in this case, utig4-100 and utig4-495). We then fill the gaps with the estimated number of loops, along with the flanking new gaps. This allows us to build an assembly that may not be perfect but is gradually improving. Additionally, multiple loops could enhance the read alignment in future use.
The most frequent number of loops in the ONT path spanning the loop node and the haplotype-assigned nodes is 3 for this gap. So, we fill this gap with 3 copies of the loop node, flanked by new gaps named loop_uncertain_copy. Additionally, for this case, we add a gap at the end of the new path, which could lead to an “unmatch” warning in the report. However, this is intentionally done, so we can ignore the warning here.
obj = vf.pp.fillGaps(obj=obj,
gapId='gapid_2',
final_path="utig4-100+ ,utig4-2421+,utig4-2421+,utig4-2421+")
final path : >utig4-100>utig4-2421>utig4-2421>utig4-2421
Updated gapId gapid_2!
✅ The start node and its direction match the original node.
✅ The start node and its direction match the original node.
[= ] 1/45 gaps filled
obj = vf.pp.fillGaps(obj=obj,
gapId='gapid_23',
final_path="utig4-69+, utig4-2421+, utig4-2421+, utig4-2421+, utig4-2421+, utig4-2421+, utig4-2421+, utig4-2421+")
final path : >utig4-69>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421>utig4-2421
Updated gapId gapid_23!
✅ The start node and its direction match the original node.
✅ The start node and its direction match the original node.
[== ] 2/45 gaps filled
Homologous nodes but hint from other haplotype#
image_path = os.path.dirname(vf.__file__) + "/data/test_giraffe/fig/gapid_24.png"
display(Image(filename=image_path,width=500, height = 500))
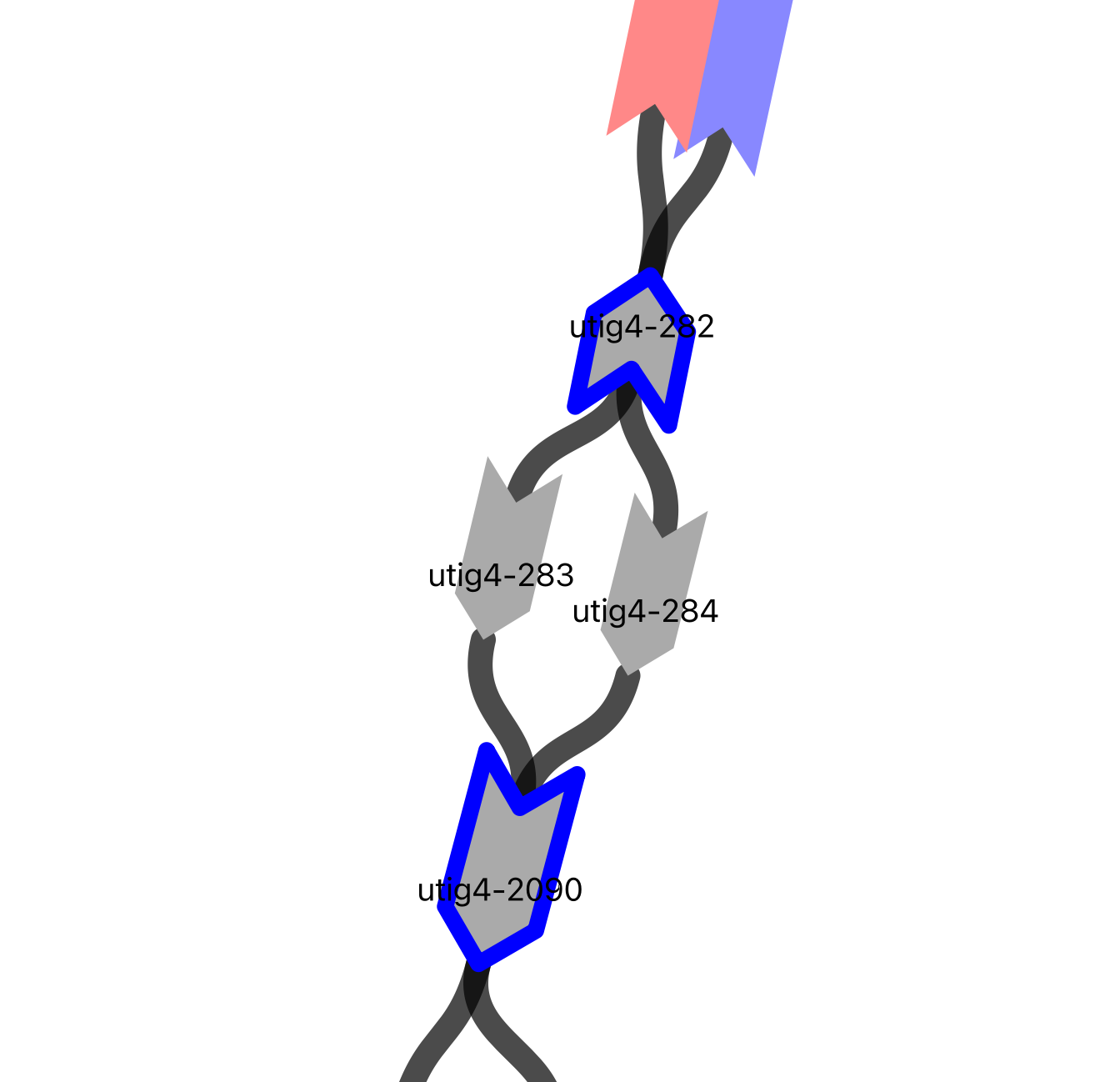
vf.pp.highlight_nodes(obj, node = "utig4-282-")
| name | path |
|---|---|
| dam_compressed.k31.hapmer_from_utig4-458 | utig4-2548+,utig4-2550+,utig4-2661-,utig4-917-,utig4-913-,utig4-914+,utig4-1121-,utig4-1123+,utig4-2641-,utig4-1381-,utig4-1379+,utig4-1383+,utig4-2677-,utig4-2586-,utig4-2584-,utig4-2484-,utig4-2482-,utig4-2027-,utig4-2023-,utig4-2024+,utig4-2601+,utig4-286-,utig4-282-,utig4-283+,utig4-2090+,utig4-810-,utig4-808+,utig4-811+,utig4-2691+,utig4-354-,utig4-353+,utig4-356+,utig4-1350-,utig4-1318-,utig4-1317+,utig4-1319+,utig4-2617+,utig4-868-,utig4-865-,utig4-289-,utig4-287+,utig4-1097+,utig4-841-,utig4-839+,utig4-52-,utig4-51+,utig4-55+,utig4-1979-,utig4-455-,utig4-454+,utig4-458+,utig4-1300-,utig4-1301+,utig4-2170+,utig4-1372-,utig4-1371+,utig4-1374+,utig4-2262-,utig4-1947-,utig4-1943-,utig4-1944+,utig4-2512-,utig4-2513+,utig4-2515+,utig4-2612-,utig4-2610-,utig4-2608+,[N5000N:ambig_path],utig4-2527-,utig4-1013-,utig4-1010-,utig4-1011+,utig4-1263-,utig4-1264+,utig4-2622+,utig4-2472-,utig4-2471+,utig4-1449-,utig4-1447+,utig4-1450+,utig4-2186-,utig4-1972-,utig4-1971-,utig4-1968-,utig4-471-,utig4-467-,utig4-468+,utig4-887+,utig4-889+,utig4-2438-,utig4-2439+,utig4-2469+,utig4-2199-,utig4-2198+,utig4-2203+,utig4-2251-,utig4-1668-,utig4-1667+,utig4-1670+,utig4-1868+,utig4-804-,utig4-800-,utig4-801+,utig4-885+,utig4-886+,utig4-768-,utig4-765-,utig4-767+,utig4-2122-,utig4-2123+,utig4-2366+,utig4-351-,utig4-348-,utig4-349+,utig4-2621+,utig4-1187-,utig4-1186+,utig4-1189+,utig4-2077-,utig4-1140-,utig4-1139+,utig4-1093-,utig4-1089-,utig4-1090+,utig4-1389+,utig4-1390+,utig4-2008-,utig4-29-,utig4-27+,utig4-30+,utig4-823+,utig4-824+ |
| sire_compressed.k31.hapmer_from_utig4-457 | utig4-2548+,utig4-2549+,utig4-2661-,utig4-916-,utig4-913-,utig4-915+,utig4-1121-,utig4-1122+,utig4-2641-,utig4-1380-,utig4-1379+,utig4-1382+,utig4-2677-,utig4-2585-,utig4-2584-,utig4-2483-,utig4-2482-,utig4-2026-,utig4-2023-,utig4-2025+,utig4-2601+,utig4-285-,utig4-282-,[N5000N:ambig_path],utig4-2090+,utig4-809-,utig4-808+,utig4-812+,utig4-2691+,utig4-355-,utig4-353+,utig4-357+,utig4-1350-,utig4-1351+,[N5000N:ambig_path],utig4-6+,utig4-14+,utig4-1317+,utig4-1320+,utig4-2617+,utig4-867-,utig4-865-,utig4-866+,utig4-869+,utig4-1097+,utig4-840-,utig4-839+,utig4-53-,utig4-51+,utig4-54+,utig4-1979-,utig4-456-,utig4-454+,utig4-457+,utig4-1300-,utig4-1302+,utig4-2170+,utig4-1373-,utig4-1371+,utig4-1375+,utig4-2262-,utig4-1946-,utig4-1943-,utig4-1945+,utig4-2512-,utig4-2514+,utig4-40-,[N5000N:ambig_path],utig4-39+,[N1000N:ambig_bubble],utig4-119-,utig4-120+,utig4-1010-,utig4-1012+,utig4-1263-,utig4-1265+,utig4-2622+,utig4-2473-,utig4-2471+,utig4-1448-,utig4-1447+,utig4-1451+,utig4-2186-,utig4-2187+,utig4-1969-,utig4-1968-,utig4-470-,utig4-467-,utig4-469+,utig4-887+,utig4-888+,utig4-2438-,utig4-2440+,utig4-2469+,utig4-2199-,utig4-2198+,utig4-2202+,utig4-2251-,utig4-1669-,utig4-1667+,utig4-1671+,utig4-1868+,utig4-803-,utig4-800-,utig4-802+,utig4-885+,utig4-511-,utig4-509+,utig4-765-,utig4-766+,utig4-2122-,utig4-2124+,utig4-2366+,utig4-352-,utig4-348-,utig4-350+,utig4-2621+,utig4-1188-,utig4-1186+,utig4-1190+,utig4-1191+,utig4-2078-,utig4-2077-,utig4-1141-,utig4-1139+,utig4-1092-,utig4-1089-,utig4-1091+,utig4-1389+,utig4-1391+,utig4-2008-,utig4-28-,utig4-27+,utig4-31+,utig4-823+,utig4-825+ |
obj.node.loc[obj.node['node'].isin(["utig4-283", "utig4-284"]), :]
| node | len | mat | pat | mat:pat | color | ont_cov | hifi_cov | |
|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
The node utig4-283 is already used in the dam haplotype, and both nodes, utig4-282 and utig4-283, have similar coverage. Therefore, we decided to include utig4-282 in the sire haplotype to fill the gap.
node_list_input = ['utig4-283', 'utig4-282']
vf.pp.searchNodes(obj, node_list_input)
Path frequency is empty, generating `obj.paths_freq`.
Filter by best mapq
Extracting paths containing nodes: ['utig4-283', 'utig4-282']
| path | utig4-283 | utig4-282 | fw | rv | total_support |
|---|---|---|---|---|---|
| <utig4-2090<utig4-283>utig4-282 | Y | Y | 87 | 87 | 174 |
| <utig4-283>utig4-282 | Y | Y | 4 | 7 | 11 |
| >utig4-282 | Y | 523 | 533 | 1056 | |
| <utig4-285<utig4-282 | Y | 98 | 85 | 183 | |
| <utig4-286<utig4-282 | Y | 93 | 88 | 181 | |
| <utig4-2090<utig4-284>utig4-282 | Y | 84 | 80 | 164 | |
| <utig4-2090<utig4-283 | Y | 9 | 6 | 15 | |
| <utig4-282>utig4-284 | Y | 3 | 3 | 6 |
Here, we can identify two clear paths: <utig4-282>utig4-283>utig4-2090 and <utig4-282>utig4-284>utig4-2090. However, we are unable to determine the haplotype using ONT alignment. In such cases, one of the obvious paths might already be used by another haplotype. Thus, we can assign the haplotype harboring this gap to the other obvious path, using an elimination method.
When we search for the utig4-282- node in the paths, the maternal (dam) strand has already used the utig4-283+ node. Therefore, we can use the utig4-284+ node to fill the gap on the paternal (sire) strand.
obj = vf.pp.fillGaps(obj=obj,
gapId='gapid_39',
final_path="utig4-282-, utig4-284+, utig4-2090+")
final path : <utig4-282>utig4-284>utig4-2090
Updated gapId gapid_39!
✅ The start node and its direction match the original node.
✅ The start node and its direction match the original node.
[=== ] 3/45 gaps filled
Homologous without hint#
image_path = os.path.dirname(vf.__file__) + "/data/test_giraffe/fig/gapid_10.png"
display(Image(filename=image_path,width=500, height = 500))
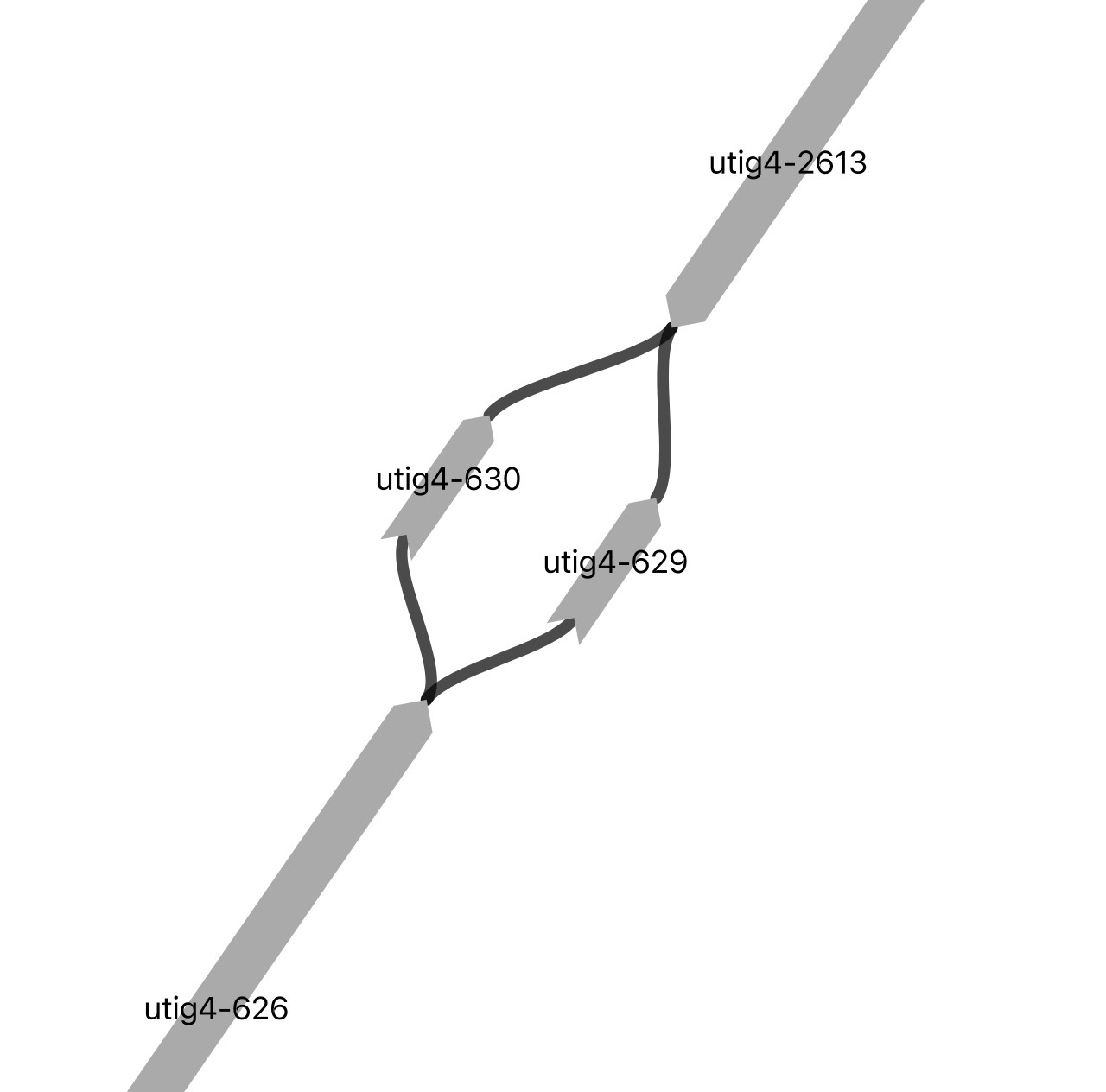
%%time
node_list_input = ['utig4-2613', 'utig4-626']
vf.pp.searchNodes(obj, node_list_input)
`obj.paths_freq` already exists.
skip generating `obj.paths_freq`.
Extracting paths containing nodes: ['utig4-2613', 'utig4-626']
CPU times: user 12 ms, sys: 1.88 ms, total: 13.9 ms
Wall time: 13.3 ms
| path | utig4-2613 | utig4-626 | fw | rv | total_support |
|---|---|---|---|---|---|
| >utig4-626>utig4-629<utig4-2613 | Y | Y | 108 | 100 | 208 |
| >utig4-2613<utig4-630<utig4-626 | Y | Y | 56 | 51 | 107 |
| >utig4-626 | Y | 4979 | 4971 | 9950 | |
| >utig4-2613 | Y | 2687 | 2637 | 5324 | |
| >utig4-612>utig4-616>utig4-2613 | Y | 110 | 82 | 192 | |
| <utig4-2613<utig4-615<utig4-612 | Y | 84 | 75 | 159 | |
| <utig4-626>utig4-628<utig4-2521 | Y | 67 | 0 | 67 | |
| <utig4-626>utig4-627<utig4-2521 | Y | 60 | 0 | 60 | |
| <utig4-627>utig4-626 | Y | 31 | 28 | 59 | |
| >utig4-626>utig4-630 | Y | 30 | 25 | 55 | |
| >utig4-2521<utig4-628>utig4-626 | Y | 53 | 0 | 53 | |
| >utig4-2521<utig4-627>utig4-626 | Y | 49 | 0 | 49 | |
| <utig4-628>utig4-626 | Y | 36 | 0 | 36 | |
| >utig4-630<utig4-2613 | Y | 20 | 13 | 33 | |
| <utig4-626>utig4-628 | Y | 24 | 0 | 24 | |
| >utig4-629<utig4-2613 | Y | 5 | 5 | 10 | |
| <utig4-2613<utig4-616 | Y | 5 | 4 | 9 | |
| >utig4-615>utig4-2613 | Y | 4 | 3 | 7 | |
| <utig4-629<utig4-626 | Y | 5 | 2 | 7 | |
| >utig4-532>utig4-2521<utig4-627>utig4-626 | Y | 2 | 0 | 2 | |
| >utig4-302>utig4-532>utig4-2521<utig4-628>utig4-626 | Y | 1 | 0 | 1 | |
| <utig4-626>utig4-627<utig4-2521<utig4-532 | Y | 1 | 0 | 1 | |
| >utig4-303>utig4-534>utig4-2521<utig4-627>utig4-626 | Y | 1 | 0 | 1 | |
| >utig4-534>utig4-2521<utig4-628>utig4-626 | Y | 1 | 0 | 1 | |
| <utig4-626>utig4-628<utig4-2521<utig4-534 | Y | 1 | 0 | 1 |
vf.pp.highlight_nodes(obj, node = "utig4-2613+")
| name | path |
|---|---|
| dam_compressed.k31.hapmer_from_utig4-1425 | utig4-857-,utig4-854-,utig4-851-,utig4-853+,utig4-2710+,utig4-2454-,utig4-2453-,utig4-59-,utig4-58+,utig4-62+,utig4-2589-,utig4-1175-,utig4-1173-,utig4-613-,utig4-612+,utig4-616+,utig4-2613+,[N1000N:ambig_bubble],utig4-626-,utig4-627+,utig4-2521-,utig4-534-,utig4-303-,utig4-301+,utig4-305+,utig4-669-,utig4-671+,utig4-928-,utig4-929+,utig4-2675+,utig4-1922-,utig4-1920+,utig4-73-,utig4-71+,utig4-74+,utig4-1424-,utig4-1425+,utig4-1845-,utig4-1847+,utig4-2685-,utig4-1587-,utig4-1584-,utig4-1585+,utig4-2457-,utig4-2050-,utig4-2049+,utig4-2052+,utig4-2291-,utig4-1197-,utig4-1193-,utig4-1195+,utig4-2194+,utig4-1865-,utig4-1863+,utig4-1867+,utig4-2382-,utig4-589-,utig4-587+,utig4-591+,utig4-1247+,utig4-1248+,utig4-2016+,utig4-1357-,utig4-1355+,utig4-1358+,utig4-2383-,utig4-2241-,utig4-2239-,utig4-1420-,utig4-1416-,utig4-1417+,utig4-1762+,utig4-1764+,utig4-2554-,utig4-2553-,utig4-2551+,utig4-1767-,utig4-1765+,utig4-1769+,utig4-2487-,utig4-963-,utig4-962+,utig4-966+,utig4-2357-,utig4-902-,utig4-898-,utig4-899+,utig4-1675-,utig4-1677+,utig4-2225+,utig4-1653-,utig4-1651+,utig4-111-,utig4-112+,utig4-2250+,utig4-1259-,utig4-1257+,utig4-1260+,utig4-2540-,utig4-2160-,utig4-2158+,utig4-2092-,utig4-2091+,utig4-2094+,utig4-2494-,utig4-2010-,utig4-2009-,utig4-1723-,utig4-1725+,utig4-1727+,utig4-2420+,utig4-1639-,utig4-1636-,utig4-1637+,utig4-2620-,utig4-1953-,utig4-1951+,utig4-1954+,utig4-2188+,utig4-1402-,utig4-1400- |
| sire_compressed.k31.hapmer_from_utig4-1426 | utig4-996-,[N34856N:tangle],utig4-856-,utig4-854-,utig4-851-,utig4-852+,utig4-2710+,utig4-2455-,utig4-2453-,utig4-60-,utig4-58+,utig4-61+,utig4-2589-,utig4-1174-,utig4-1173-,utig4-614-,utig4-612+,utig4-615+,utig4-2613+,[N1000N:ambig_bubble],utig4-626-,utig4-628+,utig4-2521-,utig4-532-,utig4-302-,utig4-301+,utig4-304+,utig4-669-,utig4-670+,utig4-928-,utig4-930+,utig4-2675+,utig4-1921-,utig4-1920+,utig4-72-,utig4-71+,utig4-75+,utig4-1424-,utig4-1426+,utig4-1845-,utig4-1846+,utig4-2685-,utig4-1588-,utig4-1584-,utig4-1586+,utig4-2457-,utig4-2051-,utig4-2049+,utig4-2053+,utig4-2291-,utig4-1196-,utig4-1193-,utig4-1194+,utig4-2194+,utig4-1864-,utig4-1863+,utig4-1866+,utig4-2382-,utig4-588-,utig4-587+,utig4-590+,utig4-1247+,utig4-1249+,utig4-2016+,utig4-1356-,utig4-1355+,utig4-1359+,utig4-2383-,utig4-2240-,utig4-2239-,utig4-1419-,utig4-1416-,utig4-1418+,[N5000N:ambig_path],utig4-1763+,utig4-2554-,utig4-2552-,utig4-2551+,utig4-1766-,utig4-1765+,utig4-1768+,utig4-2487-,utig4-964-,utig4-962+,utig4-965+,utig4-2357-,utig4-901-,utig4-898-,utig4-900+,utig4-1675-,utig4-1676+,utig4-2225+,utig4-1652-,utig4-1651+,utig4-1127-,utig4-1128+,utig4-2250+,utig4-1258-,utig4-1257+,utig4-1261+,utig4-2540-,utig4-2159-,utig4-2158+,utig4-2093-,utig4-2091+,utig4-2095+,utig4-2494-,utig4-2011-,utig4-2009-,utig4-1728-,utig4-1725+,utig4-1726+,[N5000N:ambig_path],utig4-1640-,utig4-1636-,utig4-1638+,utig4-2620-,utig4-1952-,utig4-1951+,utig4-1955+,utig4-2188+,utig4-1401-,utig4-1400- |
This scenario has the same haplotype unassignment issue as above, but there is no hint from the other haplotype. Therefore, we randomly assign the haplotype to each gap using the paths obtained from vf.pp.searchNodes(obj, node_list_input). This haplotype-switching error can be corrected during the polishing step later.
obj = vf.pp.fillGaps(obj=obj,
gapId='gapid_6',
final_path="utig4-2613+, utig4-630-, utig4-626-")
final path : >utig4-2613<utig4-630<utig4-626
Updated gapId gapid_6!
✅ The start node and its direction match the original node.
✅ The start node and its direction match the original node.
[==== ] 4/45 gaps filled
obj = vf.pp.fillGaps(obj=obj,
gapId='gapid_28',
final_path="utig4-2613+, utig4-629-, utig4-626-")
final path : >utig4-2613<utig4-629<utig4-626
Updated gapId gapid_28!
✅ The start node and its direction match the original node.
✅ The start node and its direction match the original node.
[===== ] 5/45 gaps filled
Finalize paths#
vf.pp.checkGapFilling(obj)
[===== ] 5/45 gaps filled
| gapId | name | gaps | notes | fixedPath | startMatch | endMatch | finalGaf | done | |
|---|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
The final Rukki path file to be used in the Verkko CNS run can be generated using the fixed gap file in the obj and written to the local directory for future use.
🛠️🛠️ !!! This functions will be available version v0.1.19 !!! 🛠️🛠️
Please save the obj!!
# vf.pp.writeFixedPaths(obj)
Checking for startMarker and endMarker...
Fixing paths using gap infomation...
Fixed paths were saved to assembly.fixed.paths.tsv
Fixed gaf were saved to assembly.fixed.paths.gaf
obj.history
| timestamp | activity | function |
|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
Save fillet obj#
The Verkko-fillet object can be saved as a Python pickle object using the vf.pp.save_Verkko() function. This object will be written to the Verkko output directory.
%%time
fileName = "verkko-fillet.pkl"
vf.pp.save_Verkko(obj, fileName)
save verkko fllet obj to -> verkko-fillet.pkl
CPU times: user 7.48 s, sys: 754 ms, total: 8.24 s
Wall time: 8.66 s
session_info.show()
/gpfs/gsfs11/users/kimj75/miniforge3/envs/verkko-fillet-local/lib/python3.9/site-packages/session_info/main.py:213: UserWarning: The '__version__' attribute is deprecated and will be removed in MarkupSafe 3.1. Use feature detection, or `importlib.metadata.version("markupsafe")`, instead.
mod_version = _find_version(mod.__version__)
Click to view session information
----- itables 2.2.4 pandas 2.2.3 session_info 1.0.0 verkkofillet NA -----
Click to view modules imported as dependencies
Bio 1.78 PIL 10.3.0 argcomplete NA asttokens NA cffi 1.17.1 charset_normalizer 3.4.0 colorama 0.4.6 comm 0.2.2 cycler 0.12.1 cython_runtime NA dateutil 2.9.0.post0 debugpy 1.8.11 decorator 5.1.1 exceptiongroup 1.2.2 executing 2.1.0 google NA importlib_metadata NA importlib_resources NA ipykernel 6.29.5 jedi 0.19.2 jinja2 3.1.4 kiwisolver 1.4.7 markupsafe 3.0.2 matplotlib 3.9.1 matplotlib_inline 0.1.7 mpl_toolkits NA natsort 8.4.0 networkx 3.2.1 numpy 2.0.2 packaging 24.2 parso 0.8.4 patsy 1.0.1 pexpect 4.9.0 pickleshare 0.7.5 platformdirs 4.3.6 plotly 5.24.1 prompt_toolkit 3.0.48 psutil 6.1.0 ptyprocess 0.7.0 pure_eval 0.2.3 pydev_ipython NA pydevconsole NA pydevd 3.2.3 pydevd_file_utils NA pydevd_plugins NA pydevd_tracing NA pygments 2.18.0 pyparsing 3.2.0 pytz 2024.1 scipy 1.13.1 seaborn 0.13.2 six 1.17.0 sphinxcontrib NA stack_data 0.6.3 statsmodels 0.14.4 tornado 6.4.2 tqdm 4.67.1 traitlets 5.14.3 typing_extensions NA vscode NA wcwidth 0.2.13 zipp NA zmq 26.2.0 zoneinfo NA zstandard 0.23.0
----- IPython 8.18.1 jupyter_client 8.6.3 jupyter_core 5.7.2 ----- Python 3.9.19 | packaged by conda-forge | (main, Mar 20 2024, 12:50:21) [GCC 12.3.0] Linux-4.18.0-425.19.2.el8_7.x86_64-x86_64-with-glibc2.28 ----- Session information updated at 2025-01-23 13:46
Once you save the Verkko-Fillet object locally, you can load all datasets by loading the object itself, without needing to read them individually next time.
%%time
fileName = "verkko-fillet.pkl"
obj = vf.pp.load_Verkko(fileName)
Make new verkko directory for running verkko consensus#
To run the Verkko consensus steps using the manual gap-filling path file, the first step is to generate the directory structure required for Verkko to start from the consensus step. The vf.pp.mkCNSdir function helps create this structure in the specified location by copying or creating symbolic links from the Verkko output directory. In this tutorial, we insert a new edge between ["utig4-2329","utig4-2651"] to fill the graph, so we are going to turn the missingEdge mode on.
🛠️🛠️ !!! This functions will be available version v0.1.19 !!! 🛠️🛠️
vf.pp.checkGapFilling(obj)
[=============================================== ] 45/47 gaps filled
| gapId | name | gaps | notes | fixedPath | startMatch | endMatch | finalGaf | done | cat |
|---|---|---|---|---|---|---|---|---|---|
Loading ITables v2.2.4 from the init_notebook_mode cell...
(need help?) |
Here, we finalize the object by checking the paths we want to keep and then updating the paths connected with connectContig and addContig (Please see Recovering Telomere-to-Telomere Contigs). After this step, the object is ready to generate a new folder for running the Verkko consensus step.
Note that, in this step, we ignore all nodes that are not used in the final paths where the original gaps have been closed without those nodes. As a result, these unused nodes will not be included in the new folder for running Verkko consensus and will not appear in the final assembly.
If you want to retain those nodes, you can add them to path_lst. They will then be kept during Verkko consensus steps and included in the final assembly. Similarly, if there are other contigs or nodes you want to retain, such as mitochondrial contigs that are not listed in obj.stat, please include them as well.
path_lst = ['na_unused_utig4-799', 'na_unused_utig4-798', 'na_unused_utig4-797'] # mitochondrial, which should be included.
obj, cludlst = vf.pp.finalizingVerkkoFilletObj(obj, path_lst=path_lst)
Checking for disconnected nodes and filtering out those shorter than 100000bp...
Minimum length for filtering out disconnected HPC nodes : 100000bp
Number of disconnected nodes found : 20
Number of disconnected nodes that will be filtered : 19
Disconnected paths that exceed a certain length will be preserved : 1
Keeping nodes in the unresolved gap regions...
100%|██████████| 47/47 [00:00<00:00, 46394.98it/s]
Total number of nodes used in gap filling: 0
[]
Finding paths consisting of unused nodes: 100%|█| 366/366 [00:00<00:00, 72792.23
The total number of paths consisting of nodes should be preserved.: 0
Updating the paths that are connected with connectContig and addContig...
100%|██████████| 2/2 [00:00<00:00, 14691.08it/s]
## Save the object
fileName = "verkko-fillet_gapfilled.230519.pkl"
vf.pp.save_Verkko(obj, fileName)
# obj = vf.pp.load_Verkko(fileName) # when you want to load the gapfilled object, use this line.
load verkko fllet obj from <- verkko-fillet_gapfilled.230519.pkl
path = vf.pp.writeFixedPaths(obj) # all the makred information above will be applied and this function will generate new gaf file with the new paths. you can use this for running verkko consensus.
Starting to write fixed paths to assembly.fixed.paths.tsv and assembly.fixed.paths.gaf...
The total number of original paths is 366
The number of paths that were removed:
: 300
keep_contig: 33
disconnected_node: 19
keep_Nodes_in_unresolved_gaps: 12
rm-connect-source_dam_compressed.k31.hapmer_from_utig4-1730_left_False: 1
rm-connect-source_sire_compressed.k31.hapmer_from_utig4-1859_left_False: 1
Fixing paths: 100%|██████████| 47/47 [00:00<00:00, 1284.97it/s]
gapid_42 sire_compressed.k31.hapmer_from_utig4-457 utig4-39+,[N1000N:ambig_bubble],utig4-119- not in original path
startMarker sire_compressed.k31.hapmer_from_utig4-1859 sire_compressed.k31.hapmer_from_utig4-1445 False
startMarker dam_compressed.k31.hapmer_from_utig4-1730 haplotype2_from_utig4-5 False
fix the connecting path is done!
Excluding duplicated paths: 100%|████████████| 366/366 [00:02<00:00, 162.11it/s]
The number of paths that were kept: 35
The total number of final paths is 35
Fixed paths were saved to assembly.fixed.paths.tsv
Fixed gaf were saved to assembly.fixed.paths.gaf
Using the final VerkkoFillet object, we are ready to generate a folder for running the Verkko Consensus step. The mkCNSdir function will create a folder containing all the necessary files for this step.
If you have connected contigs, set missingEdge=True; otherwise, set it to False. Also, extracting ONT sequences for the missingEdge step may take a long time.
new_folder_path = "../verkko-thic_v0.8.0_test"
vf.pp.mkCNSdir(obj, new_folder_path, missingEdge=True) # if you connect contigs, set messingEdge=True. if not, set it to False.
Creating a new verkko folder for CNS at: ../verkko-thic_v0.8.0_test
Copying the final GAF file from: assembly.fixed.paths.gaf
missing_edge_dir missing_edge is exist, set missingEdge to True
missingEdge mode is set to: True
Warning: Directory or file6-rukki does not exist in the original Verkko directory.
Handling missing edges in the new folder: /path/to/your/folder/verkko2.2_hifi-duplex_trio-hic/verkko-thic_v0.8.0_test
Found gapid list that have been filled by missing edges function : ['gapid_25', 'gapid_47', 'gapid_48', 'gapid_49']
[make_verkko_fillet_dir] Command executed successfully!
✅ All files are updated! The new folder is ready for verkko-cns.
Checking the new folder for missing files...
100%|██████████| 17/17 [00:00<00:00, 5897.70it/s]
All files exist.
Now you are ready to run Verkko Consensus using the new folder generated above!!!! You can also run Verkko Curation with this folder. Please refer to the How to Run Verkko Consensus Using a Fixed Path section in the documentation for the next steps. This is expected folder structrue and files.
.
├── 1-buildGraph -> /mypath/myVerkkoDir/1-buildGraph
├── 2-processGraph -> /mypath/myVerkkoDir/2-processGraph
├── 3-align -> /mypath/myVerkkoDir/3-align
├── 3-alignTips -> /mypath/myVerkkoDir/3-alignTips
├── 4-processONT -> /mypath/myVerkkoDir/4-processONT
├── 5-untip -> /mypath/myVerkkoDir/5-untip
├── 6-layoutContigs
│ ├── combined-alignments.gaf
│ ├── combined-edges.gfa
│ ├── combined-nodemap.txt
│ ├── consensus_paths.txt
│ ├── createLayout.err
│ ├── createLayoutInputs.err
│ ├── createLayoutInputs.sh
│ ├── createLayout.sh
│ ├── gaps.txt
│ ├── hifi.alignments.gaf
│ ├── nodelens.txt
│ ├── ont.alignments.gaf
│ └── ont-gapfill.txt
├── 7-consensus
│ ├── ont_subset.fasta.gz
│ ├── ont_subset.fasta.gz.fai
│ ├── ont_subset.fasta.gz.gzi
│ └── ont_subset.id
├── assembly.colors.csv -> /mypath/myVerkkoDir/assembly.colors.csv
├── assembly.homopolymer-compressed.noseq.gfa -> /mypath/myVerkkoDir/assembly.homopolymer-compressed.noseq.gfa
└── hifi-corrected.fasta.gz -> /mypath/myVerkkoDir/hifi-corrected.fasta.gz